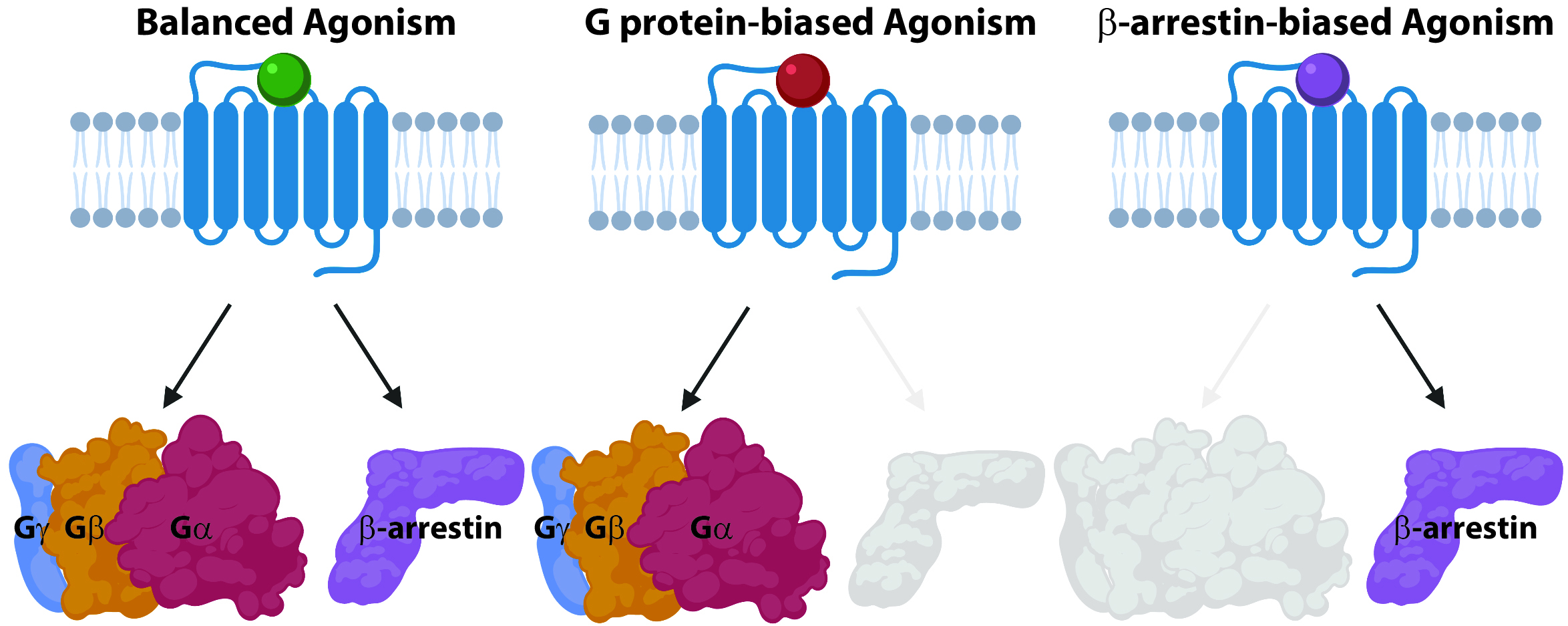
Our main research focus is on identifying novel mechanisms of receptor signaling and novel drug targets for the treatment of pulmonary arterial hypertension (PAH), a disease of the pulmonary vasculature that causes right heart failure. Today, nearly a third of FDA-approved drugs target G protein-coupled receptors (GPCRs), which are the most common receptors in the human genome and are involved in nearly every aspect of human physiology. In PAH, we use drugs that target endothelin receptors and the prostacyclin receptor. These GPCRs regulate cardiovascular physiology through the control of distinct signaling pathways, such as those regulated by heterotrimeric G proteins or β-arrestin adapter proteins. For decades GPCRs were thought to act as “on-off” switches, where agonist binding would result in an “on” state, while an antagonist would result in an “off” state. However, this model was shown to be an oversimplification with the discovery of “biased agonism.” Biased agonism is the ability of different ligands to selectively activate some pathways downstream of a GPCR, such as G proteins or β-arrestins, while not activating others (Figure 1). Biased agonism heralds an era of more precise drugs that deliver therapeutic benefits while minimizing off-target effects. Biased agonists could revolutionize our approach to targeting GPCRs, allowing us to target previously poor candidates (due to side effects), and providing the opportunity to target novel signaling pathways that could be selectively activated or inhibited. However, little is known regarding the mechanisms underlying biased agonism and the multiple pathways of arrestin-mediated signaling that these drugs target.
The overall goals of our current research program are to: 1) identify the biochemical mechanisms that underlie biased agonism and other noncanonical GPCR signaling ; 2) β-arrestin-mediated signaling in the vasculature; and 3) translational research in pulmonary vascular disease.
1. Novel Mechanisms of G protein-coupled receptor (GPCR) signaling
Physiology of Biased Agonism in Chemokine Receptors
In studying biased agonism, our lab has focused on signaling by the chemokine system, which controls immune cell function and inflammation. Across a number of vascular diseases, chemokine receptors regulate inflammation through their modulation of immune cell function. The chemokine system consists of twenty receptors and nearly fifty ligands, where most receptors bind multiple ligands and most ligands bind multiple receptors. For example, the chemokine receptor CXCR3 is expressed on macrophages and activated T cells and contributes to chronic inflammation in a number of cardiovascular diseases.
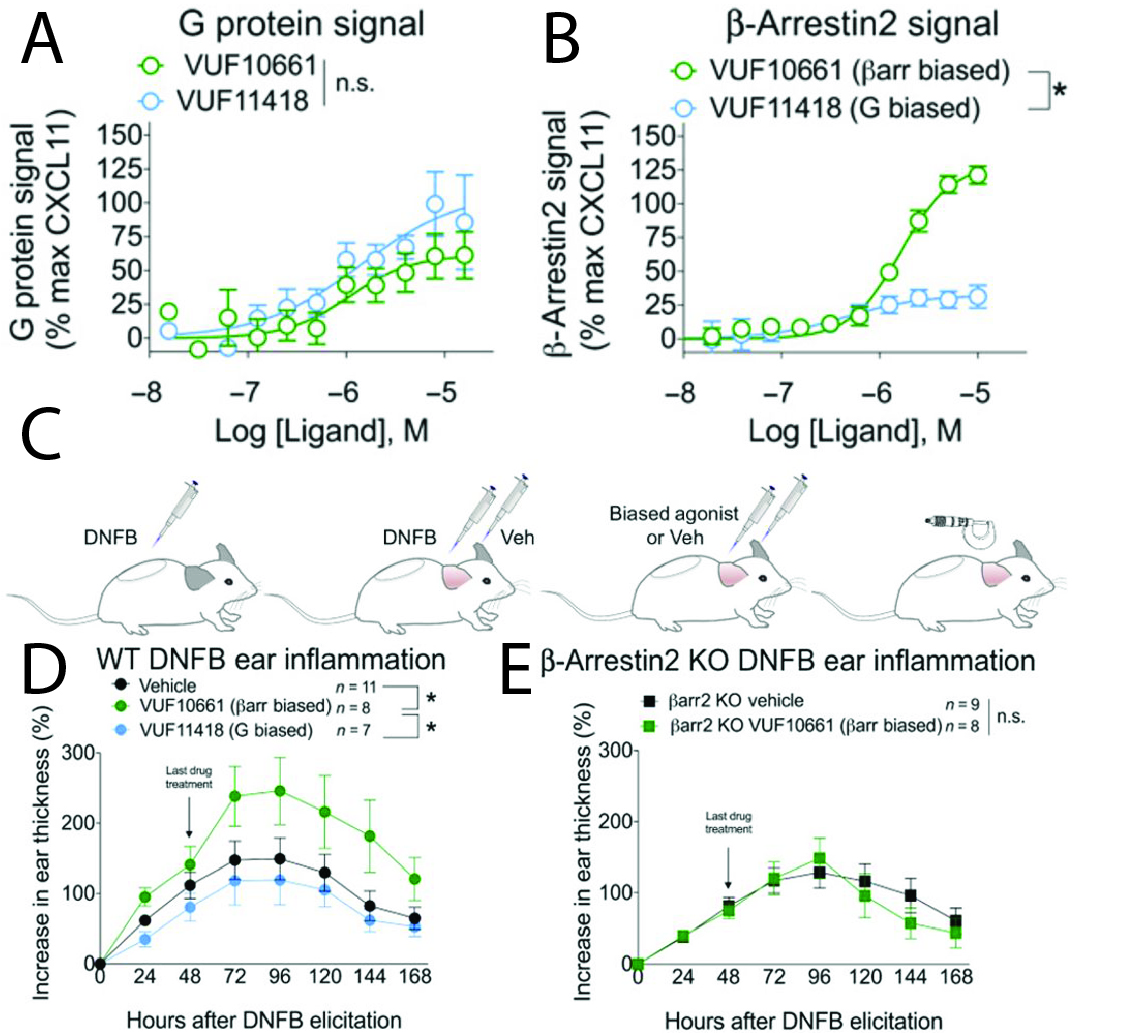
The chemokine receptor CXCR3 plays a central role in inflammation by mediating effector/memory T cell migration in vascular inflammation. However, drugs targeting CXCR3 and other chemokine receptors have largely been ineffective in treating inflammation in clinical trials. We hypothesized that this disconnect between the important role of the chemokine system in inflammation in basic science studies and the lack of efficacy of targeting the chemokine system in clinical trials was due to a knowledge gap of the physiological effects of biased chemokine signaling. Our group had shown that the three chemokines that bind CXCR3, CXCL9, 10 and 11, act as biased agonists, with CXCL11 promoting increased β-arrestin recruitment and receptor internalization compared to CXCL9 and 10. However, it was unclear how this bias translated to distinct physiological effects in inflammation. As it is difficult to use chemokines, which are 8-10 kDa in size with complex binding properties (such as binding glycosaminoglycans), in physiological experiments, we first had to identify small-molecule biased agonists that could be used in animal experiments. Taking advantage of a series of compounds that had previously been developed, we identified and characterized small-molecule biased agonists of the receptor (Fig. 2A and B). We found that VUF10661 and VUF11418 both activated G protein (Gαi) signaling but VUF10661 was β-arrestin-biased relative to VUF11418, which was relatively G protein-biased. Both of these molecules were ~ 500-700 kDa in size, allowing us to use them topically in a mouse model of T cell-mediated allergic contact hypersensitivity (CHS). In the CHS model, mice are first sensitized by application of the small-molecule hapten dinitrofluorobenzene (DNFB) to the back of the mouse (Fig. 2C). Two days later, an inflammatory reaction is elicited by DNFB treatment of the ears along with treatment with vehicle or the G protein- (VUF11418) or β-arrestin-biased (VUF10661) agonist. Inflammation was then assessed every 24 hours by measuring ear thickness with calipers. We found that topical application of the β-arrestin-biased, but not the G protein-biased, agonist potentiated inflammation (Fig. 2D), an effect that was lost in β-arrestin 2 KO mice (Fig. 2E – β-arrestin 2 is the predominant isoform in T cells). Notably, we observed that T cell recruitment was increased by the β-arrestin-biased agonist, and patient biopsies of allergic CHS demonstrated coexpression of CXCR3 and β-arrestin in T cells. Analysis of phosphorylated proteins in human lymphocytes showed that β-arrestins activated the kinase Akt, which was required for T cell migration. This research demonstrated that a β-arrestin-biased CXCR3 agonist can promote inflammation, suggesting distinct roles for different endogenous chemokines that signal through CXCR3. This also provided evidence that biased signaling can affect the clinical utility of drugs targeting chemokine receptors.
Systems Biology of Biased Signaling
While our previous studies have demonstrated that CXCR3 biased agonists can have distinct effects on inflammation, it is still unclear how chemokine biased agonists differentially activate signaling pathways that results in distinct immune responses. To address this question at a systems level, we are now performing and analyzing global phosphoproteomics and transcriptomics datasets to identify signaling pathways regulated by CXCR3 in response to stimulation by CXCL9, 10 and 11. To perform phosphoproteomics studies, we are collaborating with Drs. Jon Jacobs and Tujin Shi at Pacific Northwest National Labs (PNNL), who have developed a pipeline for highly sensitive phosphoproteomics analysis. For our initial phosphoproteomic experiments, we have had to use CXCR3-overexpressing HEK293 cells to obtain sufficient sample for analysis, while for transcriptomic studies we have used activated human donor T cells (Fig. 3A). In the phosphoproteomic dataset consisting of ~ 60,000 phosphopeptides corresponding to ~4,000 proteins, we found ~1,500 peptides were significantly different across treatment conditions (Fig. 3B), including proteins involved in cytoskeletal organization, metabolism and proliferation. Similarly, in our transcriptomic dataset (Fig. 3C), we identified differential regulation of pathways including endocytosis, leukocyte transendothelial migration and actin organization.

Gai:β-arrestin complex formation
G protein- and β-arrestin-mediated signaling have broadly been considered separable, and functional interactions between Gα proteins and β-arrestins have previously not been appreciated. Here we show GPCRs promote a direct interaction between Gαi protein subtype family members and β-arrestins, even downstream of non-Gαi coupled GPCRs. Gαi:β-arrestin complexes could bind ERK MAP kinase and their disruption impaired both ERK activation and cell migration, consistent with β-arrestins requiring a functional interaction with Gai for certain signaling events. These results introduce a new GPCR signaling mechanism distinct from canonical G protein activation where GPCRs cause the formation of Gαi:β-arrestin signaling complexes.

2. Beta-Arrestin-Mediated Signaling in the Vasculature
Beta-arrestins are ubiquitous adapters that regulate GPCR signaling, trafficking and desensitization. Although their ability to mediate signaling through kinases and other pathways has been appreciated for over a decade, the full spectrum of their activity is not fully appreciated, especially as it relates to their physiologic effects and their regulation of non-GPCR targets, such as receptor tyrosine kinases (RTKs). It is critical to link the pharmacology of these receptors to their intracellular targets and subsequent physiologic effects.
Recently, we discovered a role for β-arrestin-mediated signaling at RTKs in the pulmonary vasculature in PAH. Receptor signaling is central to endothelial function and is dysregulated in vascular diseases such as atherosclerosis and PAH. Receptors involved in endothelial function include RTKs (such as vascular endothelial growth factor receptors) and GPCRs, which classically activate distinct intracellular signaling pathways from each other. The mechanisms that regulate these signaling pathways have not been fully elucidated and it is unclear what nodes for cross talk exist between them. Using human samples from PAH lungs, we discovered that β-arrestin 1, but not β-arrestin 2, levels were significantly reduced in PAH (Fig. 5A-C). Consistent with this, we found that β-arrrestin 1 KO mice were more susceptible to hypoxia-induced PAH; after 4 weeks of exposure to hypobaric hypoxia (corresponding to an FiO2 of 0.10), β-arrestin 1 KO mice displayed significantly more RV hypertrophy (Fig. 5D), higher RV systolic pressures (Fig. 5E) and decreased RV function as assessed by RV fractional area change from time-resolved microCT measurements (Fig. 5F-G). While we initially hypothesized that these changes were due to defects in GPCR signaling, RNA sequencing of the lungs from the WT and β-arrestin 1 KO mice demonstrated that the pathway that was most significantly inhibited in β-arrestin 1 KO mice in hypoxic conditions was VEGFR3 signaling. We confirmed this in pulmonary microvascular endothelial cells, in which VEGF-C stimulation resulted in VEGFR3 phosphorylation, an effect that was lost with β-arrestin knockdown (Fig. 5H). Notably, Akt phosphorylation was also lost with β-arrestin-1, but not β-arrestin-2, knockdown (Fig. 5H), demonstrating that the β-arrestin isoforms play distinct roles in promoting VEGFR3 signaling. In additional biochemistry experiments, we found that β-arrestin-1 interacts with VEGFR3 and regulates its internalization, resulting in a model for β-arrestin 1 regulation of VEGFR3 signaling (Fig. 5I). Notably, expression of a constitutively active Akt mutant rescued the endothelial tube formation defect of pulmonary endothelial cells after β-arrestin 1 knockdown, demonstrating the physiological importance of β-arrestin-1-mediated VEGFR3 signaling via Akt. Our results demonstrated a novel role for β-arrestin 1 in VEGFR3 regulation and suggested a mechanism for cross talk between GPCRs and VEGFRs in PAH.

3. Translational Research in Pulmonary Vascular Disease
Chronic thromboembolic pulmonary hypertension (CTEPH) is a late sequela of pulmonary embolism that occurs in approximately 2% of patients with acute PE. In CTEPH, the acute PE does not resorb but instead develops into an organized “thrombus” (Fig. 6A), leading to persistent obstruction of the pulmonary vasculature, with progressive right heart failure and death. The gold standard treatment for CTEPH is pulmonary thromboendarterectomy (PTE), in which the thrombus is surgically removed from the pulmonary arterial system. However, many patients are not surgical candidates or do not have surgical disease. For these patients, novel approaches such as balloon pulmonary angioplasty or a single FDA-approved therapy can be used. Both of those interventions have only been shown to improve short-term endpoints with no long-term outcome data. Thus, the development of novel medical therapies for CTEPH is a significant unmet need. However, CTEPH drug development has been limited by a lack of understanding of the pathology of human disease and a dearth of relevant disease models. Previous studies have primarily used histopathology or cell culture from CTEPH thrombus, which are inherently limited to specific markers or to those cells that grow under cell culture conditions, respectively. To directly address this limitation, we have developed an approach to assess the heterogeneity of the thrombus with single cell RNA sequencing (scRNAseq). Using thrombus samples from five patients who underwent PTE surgery, we have develop a cell atlas of CTEPH thrombus (Fig. 6B). This has allowed us to identify distinct cell types, such as smooth muscle cells, endothelial cells, macrophages and T cells, that form CTEPH thrombus.

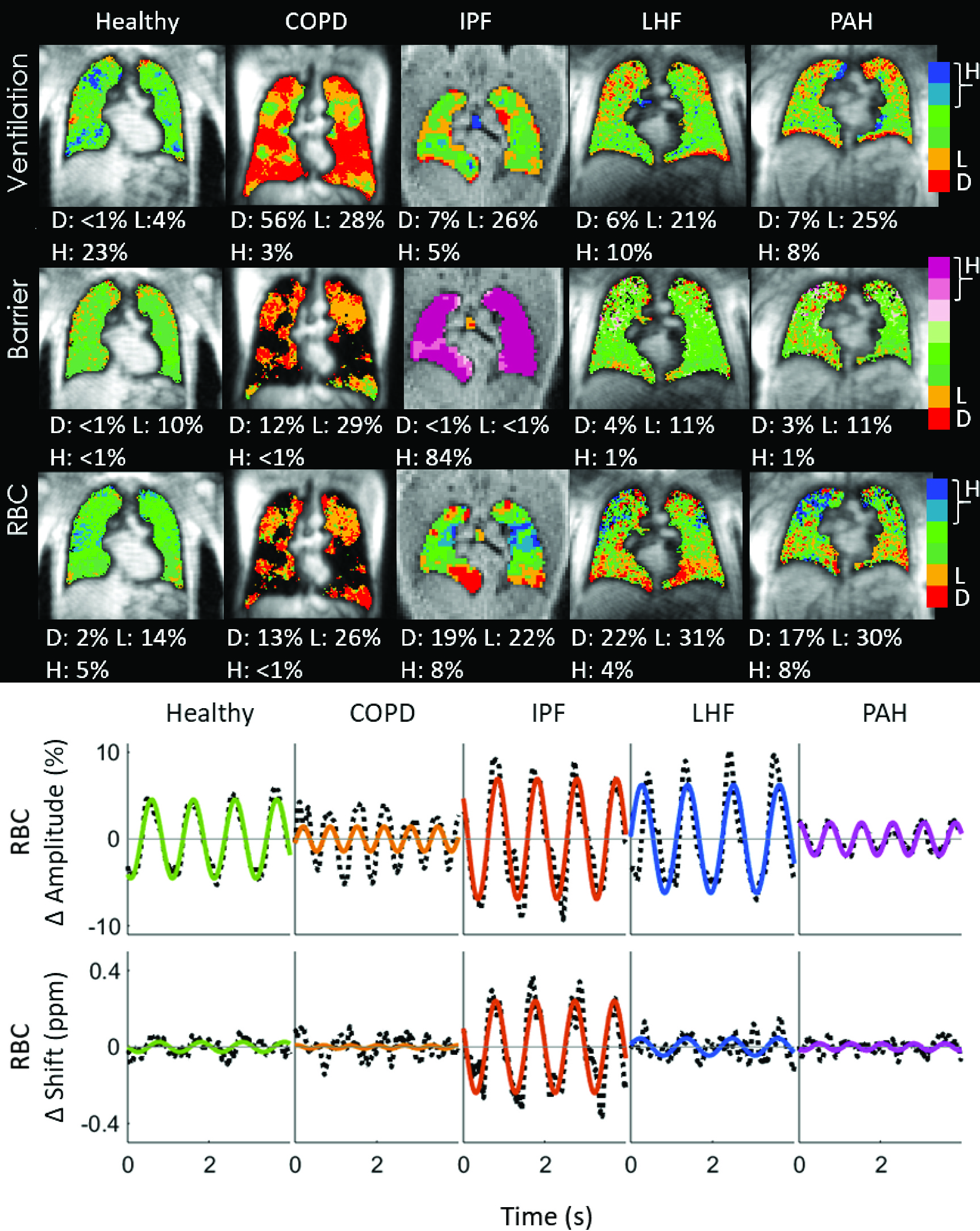
A major limitation in the assessment of PAH is a lack of specific disease activity biomarkers. For example, in a disease such as HIV, we can measure HIV viral load or CD4 count, while in many cancers we can assess tumor burden noninvasively with serum biomarkers or imaging. The assessment of PAH severity usually relies on a combination of invasive hemodynamics, functional assessment (such as six minute walk distance), serum biomarkers such as NTproBNP and imaging of the right ventricle with echocardiography or MRI. However, none of these markers are specific for PAH and none of them directly report on lung pathology. Therefore, disease-specific biomarkers are a significant unmet need in the diagnosis and management of PAH. In collaboration with Dr. Bastiaan Driehuys (Department of Radiology, Duke University), we have used hyperpolarized 129Xenon magnetic resonance imaging (Xe MRI) to develop an imaging biomarker for abnormal gas exchange in pulmonary vascular disease. In Xe MRI, hyperpolarized Xe give unique signals depending on whether it is in airspace (ventilation), lung tissue (barrier) or red blood cells (RBC – bound to hemoglobin), allowing us to visualize gas exchange physiology in a single 8 second breath-hold. The changes in the RBC spectroscopic signal also report on changes in pulmonary capillary blood volume, which is sensitive to precapillary PH. In our recently published study, we have demonstrated that PAH patients demonstrate a unique Xe MRI signature compared to other chronic heart and lung diseases, such as COPD, IPF and left heart failure (Fig. 7). Specifically, PAH demonstrates RBC defects, consistent with abnormal pulmonary capillary blood flow (Fig. 7, upper panel), along with decreased cardiogenic oscillations on dynamic spectroscopy that are associated with precapillary resistance to blood flow (Fig 7, lower panel). We believe that this technology may be useful in the diagnosis and monitoring of PAH, but some of its greatest impact may be in its application to the noninvasive monitoring of disease activity in preclinical models of PAH that can be correlated directly with human disease.