
Consideration for updating pipleline
Reply
Here are some suggestions for updating our pipeline:
1. Shall pass in the parameter for major programs as well as the parameter setting, i.e. BWA, Samtools, etc. Should return “null” for those can be null
2. For BWA samse/pe procedure, need to add “-r” option for adding the read group information.
3. Should consolidate codes (bash and tcsh should be merged) and use subversion control on version
4. Validate all input, i.e. run_summary information from core facility. There was a version issue with our exiting pipeline . Mixture of pipeline version and sample_info file.
5. We shall document our “pipeline” with version
6. Mixture of different codes: perl, bash, tcsh, etc. These need to be documented.
7. To many sub-directories, hard to manage and keep track.
8. Too many copy and paste, which can potentially introduce errors. Need to eliminate these steps and create log for each step.
9. A database/lims will be helpful for storing all these information.
How we may do better
Part I, Version control on all third party software, BWA, samtools, Picard, GATK, etc. Version control on “pipeline” itself. Goal: easy to manage, migrate and update. Modularize and easy for plugging in.
Part II, Subversion control on all codes, cleaning up code, and consolidate of mixture codes
Part III, Implement (if not) ASPERA for file transferring for anything for data management
Part IV, Error checking/QC system
Part V, In-house database and LIMS to replace Excel spread sheet, for sample tracking. In-house database for keep log for any analysis. Web programming helper needed.
Protected: GATK — on GRC37 (trouble shooting)
SVA project
This is my first time to use SVA, for recalibration project.
Project URL: Sequence Variant Analyzer
A sample .gsap file is looked like the following:
############################################################
#SequenceVariantAnalyzer project file.
#Authors: Dongliang Ge & David B. Goldstein
#Duke Institute for Genome Sciences & Policy, Center for Human Genome Variation
#Note: lines after the commenting symbol (#) will be ignored in the analysis.
############################################################
#Output file
[OUTPUT]=/nfs/svaprojects/vcf/genome/vcf_mchd002A2_recal.sva
[PEDINFO]=/nfs/svaprojects/master.ped
[COLLECTNONCARRIERINFO]=OFF
#######################################################################
#The following section lists the inputs and outputs used in the annotation. You need to specify your files.
#######################################################################
#Whole-exome sequence samples#
[INPUT]=mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/var_flt_vcf.snp.filtered.vcf
[COVERAGE]=1,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.1.bco
[COVERAGE]=2,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.2.bco
[COVERAGE]=3,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.3.bco
[COVERAGE]=4,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.4.bco
[COVERAGE]=5,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.5.bco
[COVERAGE]=6,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.6.bco
[COVERAGE]=7,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.7.bco
[COVERAGE]=8,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.8.bco
[COVERAGE]=9,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.9.bco
[COVERAGE]=10,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.10.bco
[COVERAGE]=11,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.11.bco
[COVERAGE]=12,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.12.bco
[COVERAGE]=13,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.13.bco
[COVERAGE]=14,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.14.bco
[COVERAGE]=15,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.15.bco
[COVERAGE]=16,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.16.bco
[COVERAGE]=17,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.17.bco
[COVERAGE]=18,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.18.bco
[COVERAGE]=19,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.19.bco
[COVERAGE]=20,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.20.bco
[COVERAGE]=21,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.21.bco
[COVERAGE]=22,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.22.bco
[COVERAGE]=X,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.X.bco
[COVERAGE]=Y,mchd002A2,/nfs/svaprojects/vcf/genome/mchd002A2/bco/mchd002A2_.Y.bco
[HMMSVMINLOD]=0
###########################################################################
#The following section lists an genome buffering option. Enabling this option will speedup the loading an
#integrated genome browser in this software. You will need to specify a file to store the buffered information.
###########################################################################
#Below is an example of a buffering file:
#[GenomeBrowserBuffer]=E:projectsSequenceVariantAnalyzerdatachr6smallTest.gbb
[GenomeBrowserBufferSwitch]=on
[GenomeBrowserBuffer]=/local2/dg48/projects/sequencing/gbb/cns.gb2
##################################################################
#The following section lists the annotation options.
##################################################################
#If specified as “on”, this option tells SequenceVariantAnalyzer to skip the annotation procedures
# and directly load the binary output instead.
[SKIPANNOTATION]=on
#Define upstream/downstream span
[UPSTREAM]=1000
[DOWNSTREAM]=1000
#Define intronic exon boundary. Intronic SNPs located within specified number of bases from
# exon boundary will fall into this category.
#This number has to be greater than 2.
[INTRON_EXON_BOUNDARY_INTO_INTRON]=8
[INTRON_EXON_BOUNDARY_INTO_EXON]=3
#Define threshold to group structural variations with recorded DGV CNVs – defualt is 50%.
[DGVCNVOVERLAP]=0.50
#Specify comprehensive or abbreviated annotation output. Note: comprehensive annotations include functionality for all relevant transcripts,
#while abbreviated output only includes the potentially most important one, but at a risk of losing information.
[DETAILED_OUTPUT]=on
############################################################################
#The following section lists the databases used in the annotation. You do not need to change them unless you
# need to use other versions of the databases.
############################################################################
#Reference database version
[COREVERSION]=50_36l
#Reference sequence, could be multiple lines, binary
[REFBIN]=1,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.1.fa.bin
[REFBIN]=2,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.2.fa.bin
[REFBIN]=3,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.3.fa.bin
[REFBIN]=4,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.4.fa.bin
[REFBIN]=5,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.5.fa.bin
[REFBIN]=6,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.6.fa.bin
[REFBIN]=7,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.7.fa.bin
[REFBIN]=8,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.8.fa.bin
[REFBIN]=9,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.9.fa.bin
[REFBIN]=10,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.10.fa.bin
[REFBIN]=11,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.11.fa.bin
[REFBIN]=12,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.12.fa.bin
[REFBIN]=13,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.13.fa.bin
[REFBIN]=14,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.14.fa.bin
[REFBIN]=15,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.15.fa.bin
[REFBIN]=16,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.16.fa.bin
[REFBIN]=17,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.17.fa.bin
[REFBIN]=18,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.18.fa.bin
[REFBIN]=19,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.19.fa.bin
[REFBIN]=20,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.20.fa.bin
[REFBIN]=21,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.21.fa.bin
[REFBIN]=22,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.22.fa.bin
[REFBIN]=X,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.X.fa.bin
[REFBIN]=Y,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.Y.fa.bin
[REFBIN]=MT,./datasource/ensembl/50_36l/Homo_sapiens.NCBI36.50.dna.chromosome.MT.fa.bin
#RepeatMasker result folder
[REPEATMASKERFOLDER]=./datasource/repeatMasker
#A switch to turn on/off function for generating ‘N’ bases in reference sequence
[REGENERATEREFNCALLS]=OFF
#The following are the annotation databases
#Core
[GENEID]=./datasource/ensembl/50_36l/gene_stable_id.txt
[TRANSCRIPT]=./datasource/ensembl/50_36l/transcript.txt
[TRANSCRIPTID]=./datasource/ensembl/50_36l/transcript_stable_id.txt
[TRANSLATION]=./datasource/ensembl/50_36l/translation.txt
[EXON]=./datasource/ensembl/50_36l/exon.txt
[EXONTRANSCRIPT]=./datasource/ensembl/50_36l/exon_transcript.txt
[XREF]=./datasource/ensembl/50_36l/xref.txt
[OBJECTXREF]=./datasource/ensembl/50_36l/object_xref.txt
[GENE]=./datasource/ensembl/50_36l/gene.txt
[SEQREGION]=./datasource/ensembl/50_36l/seq_region.txt
#GO
[GO]=./datasource/ensembl/50_36l/term.txt
#Existing variations
[VARIATION]=./datasource/ensembl/50_36l/variation.txt
[VARIATIONFEATURE1]=./datasource/ensembl/50_36l/variation_feature.001.txt
[VARIATIONFEATURE2]=./datasource/ensembl/50_36l/variation_feature.002.txt
[VARIATIONALLELE1]=./datasource/ensembl/50_36l/allele.001.txt
[VARIATIONALLELE2]=./datasource/ensembl/50_36l/allele.002.txt
[VARIATIONSAMPLE]=./datasource/ensembl/50_36l/sample.txt
#Protein sequence variation annotation (pre-calculated)
[PROTEINVAR]=./datasource/proteinpoly/mapp.out
#Hapmap
[HAPMAP]=./datasource/hapmap/hapmap_r23a.bim
#Illumina 1M chip
[ILLUMINA1M]=./datasource/illumina/Human1Mv1_snptable.txt
#CNV
[DGVCNV]=./datasource/CNV/DGV/variation.hg18.v3.txt
[CNVTAGGING]=./datasource/CNV/tagging/CNV_tagging_SNPs.txt
#KEGG pathway files
[KEGGMAPTITLE]=./datasource/kegg/Aug2508/map_title.tab
[KEGGGENEPATHWAY]=./datasource/kegg/Aug2508/genes_pathway.list
#Venter’s variants
[VENTERSNPS]=./datasource/venter/HuRef.InternalHuRef-NCBI.snp.bin
[VENTERINDELS]=./datasource/venter/HuRef.InternalHuRef-NCBI.indel.bin
#1000 Human Genome SNP
[1000HUMANGENOMESNP]=./datasource/1kgenome/CEU.frequency.filter
#OMIM Gene map
[OMIMGENEMAP]=./datasource/omim/genemap
#An *optional* protein sequence output
#[PROTEINFASTAOUTPUTFOLDER]=./datasource/ensembl/50_36l/proteinfasta
#######################################################
#End of the SequenceVariantAnalyzer project script file.
#######################################################
GATK assessment
This is the assessment on GATK recalibration, using our current variant calling pipeline.
Samples aligned to build36
dukscz0106
mchd002A2
na12878
Major steps:
Step 1: calling variants with samtools1.0.12a
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b36.sh
2. Input: /nfs/chgv/seqanalysis2/GATK/recalBam/dukscz0106.build36.recal.noRG.bam
3. OutputDir: /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/
4. Submit job: qsub /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b36.sh /nfs/chgv/seqanalysis2/GATK/recalBam/dukscz0106.build36.recal.noRG.bam /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/
5. Sent @7:50 a.m. June 11th, but the queue was very long!For mchd002A2: qsub /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b36.sh /nfs/chgv/seqanalysis2/GATK/recalBam/mchd002A2.build36.recal.noRG.bam /nfs/chgv/seqanalysis2/GATK/samples/mchd002A2/noRG_recal/
For na12878, run on lsrc and results were transferred back to dscr.
For comparison purposed, run one on the dscr as well @1:55 p.m. June 23rd, 2010: qsub /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b36.sh /nfs/chgv/seqanalysis2/GATK/recalBam/na12878.build36.recal.noRG.bam /nfs/chgv/seqanalysis2/GATK/samples/na12878/noRG_reCal/
Here are a few QC metrics we can check
1. ti/tv ratio
2. number of raw and filtered SNV
3. number of indel called by samtools
4. proportion of heterozygous SNVs on the x chr compared to the total number of SNVs in the X
5. Ratio of homo to hetero SNVs
Split vcf files by snps and indels
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl
2. Input: /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/var_flt_vcf (var_raw_bcf is binary file, where var_flt_vcf was generated from), also, this “var_flt_vcf” is really “RAW” SNV/INDEL file.
3. RunningDir/OutputDir: /nfs/chgv/seqanalysis2/GATK/runningDir
4. cd to the RunningDir, and submit job:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl /nfs/chgv/seqanalysis2/GATK/runningDir/dukscz0106_vcf_files , this file contains a var_flt_vcf file to process.dukscz0106 /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/var_flt_vcf
5. For mchd002A2: qsub /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl /nfs/chgv/seqanalysis2/GATK/runningDir/mchd002A2_vcf_files
6. For na12878: qsub /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl /nfs/chgv/seqanalysis2/GATK/runningDir/na12878_vcf_files
7. For na12878_raw: qsub /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl /nfs/chgv/seqanalysis2/GATK/samples/na12878/no_reCal/na12878_noGATK_vcf_files
Filter vcf variants
/nfs/chgv/seqanalysis/ALIGNMENT/QC/Scripts/sam12/vcf_varfilter.pl
Your variant list should be in the format;
Two files with the same name as your variant file will be created:
.filtered (variants that passed QC threshold)
.filtered.out (variants which failed QC)
Example usage:
qsub /nfs/chgv/seqanalysis/ALIGNMENT/QC/Scripts/sam12/vcf_varfilter.pl /nfs/chgv/seqanalysis/ALIGNMENT/QC/samtools_version/sam12/genome/vcf_snp_filelist snp
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl
2. Input: /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/var_flt_vcf.snp
3. RunningDir/OutputDir: /nfs/chgv/seqanalysis2/GATK/runningDir
4. cd to the RunningDir, and submit job:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl /nfs/chgv/seqanalysis2/GATK/runningDir/vcf_snp_dukscz0106 snp/,
vcf_snp_filelist contains a list of var_flt_vcf.snp to process.dukscz0106 /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/var_flt_vcf.snp
5. For mchd002A2: qsub /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl /nfs/chgv/seqanalysis2/GATK/runningDir/vcf_snp_mchd002A2 snp
6. For na12878: qsub /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl /nfs/chgv/seqanalysis2/GATK/runningDir/vcf_snp_na12878 snp
7. For na12878_raw: qsub /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl /nfs/chgv/seqanalysis2/GATK/samples/na12878/no_reCal//vcf_snp_na12878_raw snp
ti/tv ratio
1. Samples ( dukscz0106 & mchd002A2) located: /nfs/chgv/seqanalysis2/GATK/samples/
2. Script (gatk_titv_b36.sh) located: /nfs/chgv/seqanalysis2/GATK/scripts/
3. Log files: /nfs/chgv/seqanalysis2/GATK/logs/
4. Usage: qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv_b36.sh /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal dukscz0106.gatk_titv.csv
5. For mchd002A2: qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv_b36.sh /nfs/chgv/seqanalysis2/GATK/samples/mchd002A2/noRG_recal/ mchd002A2.gatk_titv.csv
6. For na12878: qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv_b36.sh /nfs/chgv/seqanalysis2/GATK/samples/na12878/noRG_reCal_lsrc/ na12878.gatk_titv.csv7. For na12878_raw: qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv_b36.sh /nfs/chgv/seqanalysis2/GATK/samples/na12878/no_reCal/ na12878.gatk_titv.csv
Homozygous/heterozygous SNV ratio
Check to see if the genotype is listed in the 10th column in the VCF file
Script: /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl
Usage: perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl vcf_file > printTO
Example: perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl var_flt_vcf.snp.filtered > vcf.snp.filtered.hets
Usage: cd to /workingDir/ and run the script:
perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl var_flt_vcf.snp.filtered > dukscz0106.hets
perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl var_flt_vcf.snp.filtered > mchd002A2.hets
perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl var_flt_vcf.snp.filtered > na12878.hets
perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl var_flt_vcf.snp.filtered > na12878_raw.hets
Step 2: Generate SVA project with samtools1.0.12a
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/bcftools_coverage_bco_build36.sh
2. Input(dscr): /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/file1
3. OutDir(lsrc)r: /nfs/testrun/GATK-recal/assessmentComp/samples/dukscz0106/noRG_reCal/
4. Submit job: qsub /nfs/chgv/seqanalysis2/GATK/scripts/bcftools_coverage_bco_build36.sh dukscz0106 /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/ /nfs/testrun/GATK-recal/assessmentComp/samples/dukscz0106/noRG_reCal/5. Job for mchd002A2: qsub /nfs/chgv/seqanalysis2/GATK/scripts/bcftools_coverage_bco_build36.sh mchd002A2 /nfs/chgv/seqanalysis2/GATK/samples/mchd002A2/noRG_recal/ /nfs/testrun/GATK-recal/assessmentComp/samples/mchd002A2/noRG_reCal
For dukscz0106, sent @12:05 p.m. June 11th!
For mchd002a2, sent @8:36 p.m. June 20th!
Here we can check:
6. concordance of SNVs with genotyping chip computed with SVA
Step 3. Overlap of filtered autosomal SNVs with dbsnp
1. Samples ( dukscz0106 ) located: /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/var_flt_vcf.snp.filtered
2. Script (dbsnp_overlap.pl) located: /nfs/chgv/seqanalysis2/GATK/scripts/dbsnp_overlap.pl
3. Log files: /nfs/chgv/seqanalysis2/GATK/logs/
4. Command: qsub /nfs/chgv/seqanalysis2/GATK/scripts/dbsnp_overlap.pl /nfs/chgv/seqanalysis2/GATK/doc/dukscz0106.snp
5. For mchd002A2: qsub /nfs/chgv/seqanalysis2/GATK/scripts/dbsnp_overlap.pl /nfs/chgv/seqanalysis2/GATK/doc/mchd002A2.snp
6. For na12878: qsub /nfs/chgv/seqanalysis2/GATK/scripts/dbsnp_overlap.pl /nfs/chgv/seqanalysis2/GATK/doc/na12878.snp
7. Results were written to log file
7. overlap of filtered autosomal SNVs with dbsnp
Learning from Jessica on variant call
Concordance check with SVA
(1) Generate bco files in the DSCR
Use the following 2 lines from this script
/nfs/chgv/seqanalysis2/GATK/scripts/bcftools_coverage_bco.sh:
Modify all the parameters, including Java path, samtools location, reference DB, etc.
If it runs manually, a few subdirectories need to be created properly.
a. bco_dir=$combined_dir/bco
b. $bco_dir/$sample
c. seqsata_sample_dir=/nfs/testrun/GATK-recal/assessmentComp/samples/$sample (for the results to be transferred back to our sva clusters).
Note: For mchd002A2, job sent May 2nd, @ 11:00 a.m. and finishes @ 1:02 a.m. May 3rd
Note: For dukscz0106, job sent May 3rd, @ 11:16 a.m. and finishes @ 3:30 a.m. May 4th
Usage: qsub bcftools_coverage_bco_jyl.sh /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/postCal dukscz0106
At last step, those bco files should be transferred back to LSRC
(2) Create an SVA project (meaning get a .gsap file created)
Example of SVA project:
/nfs/svaprojects/vcf/genome/vcf_genomes_3.gsap
Details see the explanation on the .gsap file
/nfs/testrun/GATK-recal/assessmentComp/samples/mchd002A2/vcf_mchd002A2_recal.gsap
/nfs/testrun/GATK-recal/assessmentComp/samples/dukscz0106/vcf_dukscz0106_recal.gsap
(3) Topstrand file names for the 2 samples:
/nfs/svaprojects/vcf/genome/topstrandfilenames (Illumina created/provided this file)
/nfs/testrun/GATK-recal/svaprojects/vcf/genome/topstrandfilenames, the content in this file is as following. The original copy was saved and individual one is used per sample.
mchd002A2, MCHD002, /nfs/sva01/SequenceAnalyses/TopStrandFiles/MCHD_Family__FEB_11_2010_TOP_FinalReport.csv
dukscz0106, Duk-Schiz0106DEF, /nfs/sva01/SequenceAnalyses/TopStrandFiles/Duke_Scz_16_samples__DEC_09_09_FinalReport_TOP.csv
(4) Use this version of SVA: (It is about 120 mitues.
/nfs/goldstein/goldsteinlab/software/SequenceVariantAnalyzer_1.1 (how to use this program??)
(Make sure to uncheck the filtering option in sva)
Step 1. Grab any machine along the hallway and log in with netID/password
Step 2. Make sure that the drive has been mounted
Step 3. cd to the directory /nfs/goldstein/goldsteinlab/software/SequenceVariantAnalyzer_1.1/, as this is the version for new VCF format
Step 4. Lunch SVA, ./SequenceVariantAnalysis.sh
Step 5. From File -> Open, explore through to the directory that contains that .gsap file, make sure that all the parameters are set up properly
Step 6. Clicking “Annotating”
(5) Check concordance:
SVA version: /nfs/goldstein/goldsteinlab/software/SequenceVariantAnalyzer_1.1/
Case I: Once “annotate” is done, go directly to the following steps to get concordance.
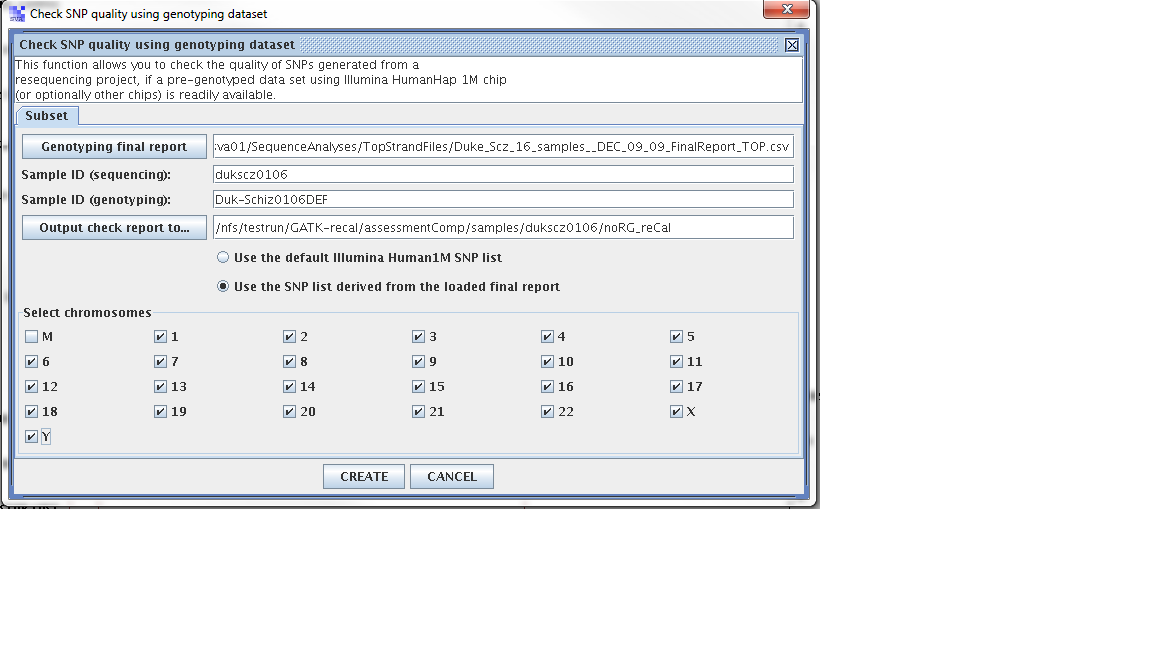
Step 1. From the SVA UI, choose “summary -> check SNP quality using genotyping dataset”
Step 2: Filling in information for four fields, information is stored at
/nfs/testrun/GATK-recal/svaprojects/vcf/genome/opstrandfilenames, the content in this file is:
mchd002A2, MCHD002, /nfs/sva01/SequenceAnalyses/TopStrandFiles/MCHD_Family__FEB_11_2010_TOP_FinalReport.csv
Four fields
Genoptying final report: /nfs/sva01/SequenceAnalyses/TopStrandFiles/MCHD_Family__FEB_11_2010_TOP_FinalReport.csv
sample ID (sequencing): mchd002A2
sample ID (genotyping): MCHD002
output check report to: /nfs/testrun/GATK-recal/assessmentComp/samples/mchd002A2
Step 3: Check “use the SNP list derived from the loaded final report”
Step 4: Click “Create”
Case II: If the project is closed, load the project by clicking “load” button and go back to the steps mentioned above.
Results:
Performing quality check using genotype data (user SNP list).
> Sample ID (sequencing): mchd002A2
Sample ID (genotyping): MCHD002
Note: 584882 SNPs are loaded from user SNP report.
– Chromosome: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X
– 300585 SNPs (with rs# listed in genotype data (user SNP list) are loaded from your resequencing project.
– 150265 SNPs on bottom strand (Illumina).
– Among these, 300351 SNPs are found in genotype data (user SNP list) .
– Among the 300585 SNPs loaded from resequencing project, there are:
299325 match with genotype data (user SNP list);
1026 mismatch;
234 not found in genotype data (user SNP list).
– Among the 1026 mismatches, there are
16 hom (seq) – hom (geno)
34 hom (seq) – het (geno)
21 het (seq) – het (geno)
955 het (seq) – hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/mchd002A2/noRG_reCal.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/mchd002A2/noRG_reCal.nomatch.txt
[Part 2] N = 283814
– Among the 283814 SNPs included in the genotyping chip but not derived from sequencing, there are:
283814 that can be correctly mapped to reference sequence;
Among these 283814 there are 280139 matches;
and 3675 mismatches, including 2798 het (geno) and 877 hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/mchd002A2/noRG_reCal.part2.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/mchd002A2/noRG_reCal.part2.nomatch.txt
For sample dukscz0106, the cooncordance check results are
Performing quality check using genotype data (user SNP list).
> Sample ID (sequencing): dukscz0106
Sample ID (genotyping): Duk-Schiz0106DEF
Note: 583754 SNPs are loaded from user SNP report.
– Chromosome: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X
– 297679 SNPs (with rs# listed in genotype data (user SNP list) are loaded from your resequencing project.
– 148586 SNPs on bottom strand (Illumina).
– Among these, 297646 SNPs are found in genotype data (user SNP list) .
– Among the 297679 SNPs loaded from resequencing project, there are:
294629 match with genotype data (user SNP list);
3017 mismatch;
33 not found in genotype data (user SNP list).
– Among the 3017 mismatches, there are
20 hom (seq) – hom (geno)
124 hom (seq) – het (geno)
88 het (seq) – het (geno)
2785 het (seq) – hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/dukscz0106/noRG_reCal.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/dukscz0106/noRG_reCal.nomatch.txt
[Part 2] N = 287131
– Among the 287131 SNPs included in the genotyping chip but not derived from sequencing, there are:
287128 that can be correctly mapped to reference sequence;
Among these 287128 there are 280499 matches;
and 6629 mismatches, including 5652 het (geno) and 977 hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/dukscz0106/noRG_reCal.part2.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/dukscz0106/noRG_reCal.part2.nomatch.txt
concordance ratio = (294629+280499)/(297646+287128) = 0.9835
QC metrics checked: comparison_to_report_JM
SVA concordance only: concordance_comparison
To compare details concordance checked done on raw bam file, all the sva project logs are located: /nfs/svaprojects/vcf/genome/concordance
For na12878 sample, we don’t have the genotype array data to compare to (as we did with in house samples). However, we do have the 1000 genome snp releases.
Step 1. Download the 1000 genome release
Step 2. Compare our in-house vcf file to the 1000 genome release.
Script located: /nfs/chgv/seqanalysis2/GATK/scripts
Script usage: qsub /nfs/chgv/seqanalysis2/GATK/scripts /dir/ /dir/
Script output: Results will be printed to log file.
Step 3. Comparison results are here: concordance_na12878
Dr. Hao Chen’s microarary project
1. Met with Dr. Hao Chen for her Microarray data analysis, the strategy to approach this request is as following:
a. Read to two references in the paper by Talor, J.M. et al 2009, PLoSOne (ref. 46 & 47) for pattern extraction analysis
b. Extract 27 possible patterns from the esophagus developing stage related genes, produce figure 2 like results in Talor’s paper
c. Will provide consultation for clustering analysis for Hao
d. Discussed the authorship for possible publication
Got it into GeneSpring GX11.0
1. With help from JP from Bioinformatics center I was able to enter the data into GeneSpring GX11.0
2. Need to create the technology first and follow the self-explanary steps
3. Create, project -> experiment
4. Do the analysis within GeneSpring
Combining data from preview study (further request from Dr. Chen, Xiaoxin)
Goal: To determine what genes (especially transcription factors) may play critical roles at each stage during esophageal development.
1. E8.25: “E8.25 definitive endoderm” will develop into future esophagus, stomach, liver, intestine… Presumably some genes should be overexpressed and some others underexpressed in oder to “specify” differentiation into E11.5 esophagus.
2. E11.5: Simple columnar epithelium, 1-2 layers
3. E15.5: Stratified squamous epithelium, 2-3 layers
4. P0: Stratified squamous epithelium, Early keratinization, 3-5 layers
5. P7: Stratified squamous epithelium, Late keratinization, 3-5 layers
Two kinds of array data are available for analysis:
1. Previous arry data: E8.25 endoderm, E11.5 esophagus (Sherwood RI, Chen TY, Melton DA. Transcriptional dynamics of endodermal organ formation. Dev Dyn. 2009 Jan;238(1):29-42). Data can be download from EO datasets (GSE13040 record). There are 3 samples of “E8.25 definitive endoderm” and 3 samples of “E11.5 esophagus endoderm” in this dataset. Illumina mouseRef-8 v2 microarrays were used in this study.
2. Hao’s array data: E11.5 esophagus, E15.5 esophagus, P0 esophagus, P7 esophagus (3 samples of each). Initially we planned to include adult esophagus. Due to our own mistakes, we decide to move data analysis forward without these samples. You can access our original data from UNC. Hao gave you SAM data. I guess after we pool E8.25 data from a previous study, we need to do analysis again.
A few issues for you to consider:
1. How to pool these two kinds of data together into a speardsheet containing 15 samples (5 time points, 3 samples each time point)?
2. SAM: Do we need to do SAM before clustering? Seems to me that clustering is good enough to show tendency of gene expression changes.
3. Hierachical clustering: Probably two categories (low and high) is enough instead of 3 categories (zero, low and high). We are interesting in genes which increase over time, or decrease over time.
4. K-means clustering: I do not know what additional information we may get from K-means.
What we would like to have in the end are:
1. A figure to show that our array data are of high quality (hierachical clustering)
2. Clustering figures to show gene expression change over time, and corresponding spreadsheets
3. Genes which define each time point
New Milestone — an email from Dr. Chen, Xiaoxin proposing divide and conquer straregy
Jianying,
Hao and I have discussed your PPT and 16 patterns. We can make some sense out of Pattern 1 to 6. We are not fully satisfied because some known critical genes (Cdx1, Cdx2, pax9) did not show up. I guess after we merged two datasets we lost a lot of data. So, let’s go for Plan B in our paper as follows:
1. We first show Agilent data (E11.5-E15.5-P0-P7, 38,290 rows).
a. patterns
b. pathways (GSA, GSEA, etc)
2. We then show Illumina data (E8.25-E11.5; 24,189 rows)
a. SAM analysis
b. pathways (GSA, GSEA, etc)
3. Finally we show E8.25-E11.5-E15.5-P0-P7 (5,652 genes)
a. merging two datasets
b. Patterns 1 to 6
For Part 1, you worked a little bit on Hao’s SAM data. It may not be good enough. You may start with original array data. For Part 2, the other group did a little bit data analysis. But their major focus was not esophageal development. Their data analyis was not optimal in finding genes/pathways which are critical for esophageal specification. For Part 3, you have finished all. Any questions please feel free to discuss.
Xiaoxin
Met with Xiaoxin, Hao, and ?
1. Adjust delta value according to q value, done but need to automate the process.
2. Get new gene list (pending on Xiaoxin’s response)
3. Fisher’s exact test (pending on Xiaoxin’s response)
4. Gene list sent out put from SAM analysis, email xiaoxin priorto down stream analysis
Pending information from hao
1. four more arrays,
2. swap sample info
Action items:
1. Talk to JP for gene spring license issue
2. GSA/GSEA analysis
3. Profiling plots…
As of May 18th,
1. Was able to have three DBs work for our GSA (I may need to explore the OVR on the Agilent data (four stages) as I am not quite confident on the alleged “mulitclass” advantage (by Tibshirani -:) in GSA (vs. GSEA)
2. Got the code worked for clustering
3. BUT, I ran into memory issues with R (on my computer). That said, just for the “viewing” and/or “detecting mislabeling”, can I may just use partial genes? ( I used to try this out whenever I ran into memory issue). But, will figure out an inclusive solution for this in the future.
Things are pending:
1. GeneSpring (have not got a chance to work on it yet)
2. Will test EPIG and make sure it works on this small laptop prior to having a more power windows server set up.
(As we all agreed, this is not critical and should not hinder us from advancing with our paper writing)
Recommendation:
Maybe we should start to layout what we have and start to draft the manuscript. I bet that we may need more (hopefully not significant more) when we start to write.
Opps! Possible mislabeling!
Thank you so much for your prompt response. Xiaoxin and I went through the treeview file and feel that we should separate the samples of 35 and 36. Would you plz cluster 35 samples and 36 samples individually? And exchange 36A1 and 35D3 before you do clustering. We will get 2 cluster fliles: one for 35 and another one for 36. We hope the results would be more organized.
Best,
Hao
Protected: A “new” pipeline with GATK recalibration
Clean up disk
It seems an issue here at CHGV on the disk space:
First of all, I need to clean up the disk on /nfs/svaprojects/ and transfer my stuffs to /nfs/testRun/
Next is /nfs/chgv/seqanalysis3/
Hi Jianying,
There’s no way you could have known this but Latasha has top priority for using seqanalysis3 and she regularly uses all of that space.
In the future, only use seqanalysis3 after consulting with Latasha.
Jessica
OK, here is the deal:
On the dscr cluster, seqanalysis3 has the highest priority for running the pipeline, so stay away from it.
Therefore, try to use /nfs/chgv/seqanalysis2/ instead
On the lsrc cluster:
I have /nfs/testrun/ directory, which has 2T space. Should be enough for now.