
Protected: Macaque genome allignment, variant call, and annotation
Protected: Build genome/exome alignment pipeline on lsrc
Protected: Help with Joe Saelens
quick&dirty shell scripts linux
Reply
I am accumulating some cool shell scripts in linux
Scenario 1
Case: I own ” /nfs/sva01/SequenceAnalyses/Concordance/BUILD37/b37_exome_concordance.csv”, so Latasha’s concordance check fails
Solution:
Step 1. Create a shortList
Step 2. Run this script:
for k in `cat shortList `; do echo $k; qsub /nfs/seqsata12/ALIGNMENT/BUILD37/EXOME/$k/Scripts/concordance.sh; done;
Scenario 2
Case: “bwa59_b37_v4.pl” has a bug, therefore symbolic link does NOT work.
Solution
Step 1: create a shortList
Step 2: run the following script,for k in `cat shortList `; do echo $k; cd /nfs/seqsata12/ALIGNMENT/BUILD37/EXOME/$k/combined ; ln -s var_flt_vcf var_flt_vcf.vcf ; cd ../../ ; done;
Scenario 3
Case: There are many columns in/nfs/sva01/SequenceAnalyses/Concordance/BUILD37/b37_exome_concordance.csv, and it is a comma-delimit files. I want a specific column
Solution
Step 1. Replacing the “,” by tab
Step 2. Use “cut -f” command
Step 3. Replacing unnecessary characters, i.e. quote, space etc.
Step 4. Grep the pattern..Run the following scripts,
sed ‘s/,/t/g’ temp.csv | cut -f 4 | more
sed ‘s/,/t/g’ temp.csv | cut -f 4 | sed ‘s/”//g’ | grep -w “NA”
Scenario 4
Case: Finding record between two files
Solution
Step 1. Samples in a file that exist in another file (intersection)
for k in `cat file1`; do grep $k file2 > /dev/null && echo $k ; done;Step 2. Samples in a file that do not exist in another file (negation)
for k in `cat file1`; do grep $k file2 > /dev/null || echo $k ; done;Step 3. Real scenario:
for k in `cat samples_wo_topstrandFile`; do grep $k /nfs/sva01/SequenceAnalyses/Concordance/BUILD37/b37_exome_concordance.csv > /dev/null && echo $k ; done;
Scenario 5
Case: There are many columns in /nfs/chgv/seqraw01/summaries/exRealign_Summary_b37.txt, and we wanted to extract “sample name information” from column three. Unfortunately, there are various number of directory layers. Also, there are duplicate sample entries.
Solution
Step 1. Use “cut -f 3 ” to extract the third column
Step 2. The split column three by “/”
Step 3. The use awk to print the last field
Step 4. Sort the list and get unique onlyWatch out for those “quote”, they many not work if directly copied to Unix/Linux environment. Special handling may be needed!
Run the following scripts,
cut -f 3 ~/exRealign_Summary_b37.tx | sed ‘s///t/g’ > allSamples.txt
awk ‘{print $NF}’ allSamples.txt > allSampleList.txt
sort -t $’t’ -k1,1 allSampleList.txt | uniq > uniqSampleList.txtOkay, in a single line:
cut -f3 /nfs/chgv/seqraw01/summaries/exRealign_Summary_v1.8_02.02.12.txt | sed ‘s///t/g’ | awk ‘{print $NF}’ | sort -t $’t’ -k1,1 | uniq | wc -l
Scenario 6
Case: There are many columns in a file and are tab-delimited, I want to know how many columns it has
Solution
For tab-delimited:
[jl407@sva no_filter]$ awk -F’\t’ ‘{print NF;exit}’ ../concordanceMeta0001.part2.match.txt
3
Okay, there are only 3 columns!!For space-delimited:
[li11@ehscmplp09/bioinfo1 RNAseqPrep]$ awk -F ‘ ‘ ‘{print NF; exit }’ refDB/UCSC_KnownGene_Dec2009/KnownGeneDec09.hg19.exon.txt
3Bingo!!
Scenario 7
Case: I have a string and want to shorten a character in the beginning and end simultaneously.
Solution
$string =~ s/^.(.*).$/$1/;
Scenario 8
Case: I have two lists, and I want to get intersection, union, and unique list.
Solution
#Getting intersection
comm -12 <(sort noMatchedTotal_inHouse) <(sort noMatch_intersect)
grep -xF -f set1 set2
sort set1 set2 | uniq -d#Getting unique list
comm -23 <(sort noMatchedTotal_inHouse) <(sort noMatch_intersect) > noMatch_uniq_inHousecomm -23 <(sort noMatchedTotal_SVA) <(sort noMatch_intersect) > noMatch_uniq_SVA
#Getting union list
sort -n set1 set2 | uniqcat set1 set2 | awk ‘!found[$1]++’
awk ‘!found[$1]++’ set1 set2
awk ‘{print $0}’ set1 set2 | sort -u
Scenario 9
Case: Working with two loops in bash search
for k in `cat ~/tmp_sample_list`; do echo $k; for f in seqsata01 seqsata02 seqsata03 seqsata04 seqsata05 seqsata06 seqsata07 seqsata08 seqsata09 seqsata10 seqsata11 ; do echo /nfs/$f/ALIGNMENT/BWA_OUTPUT; ls -l /nfs/$f/ALIGNMENT/BWA_OUTPUT | grep “$k”; done; done;
Scenario 10
Case: There was an error caused by loosing of a computer node “compute2 was shut down abruptly”. So, we had to replace all “copy.sh” scripts”. Here is the bash script Joe provided. Pretty cool!
for i in `ls -1 */Scripts/copy.sh | cut -d’/’ -f1`; do cat $i/Scripts/copy.sh | sed ‘s/compute2/sva10/g’ > $i/Scripts/newcopy.sh | mv $i/Scripts/newcopy.sh $i/Scripts/copy.sh ; done;
Please be aware that this is “-1” (one) after “ls” but not “l”
Scenario 11
Case: I have a huge text files and I wanted to strip out the first 3 lines.
To exclude the first three lines of a file
tail -n +3 file > newfileTo exclude/remove blank lines from a text file, use the following command:
$ sed ‘/^$/d’ input.txt > output.txt
$ grep -v ‘^$’ input.txt > output.txtTo remove whitespace after the ID in genomic sequence, I used this trick. It seems working pretty well.
perl -plane ‘s/\s+.+$//’ PipeIN genome.fa PipeOUT new_genome.faI have a huge text files and I wanted to strip out the last line, use the following GNU command
sed -i '$ d' foo.txt
Scenario 12
Case: I want to eliminate empty lines in a file
Solution, use “sed” to process files under linux
sed ‘/^$/d’ myFile > tt
mv tt myFile
Another useful application with “sed”, to delete lines matching the expression
sed ‘/^expression/ d’ infile.txt > outfile.txt
To parse out the “Read depth information in a VCF format file”, it becomes quite convenient
cut -f8 C3_M_fltVCF.vcf | cut -d ";" -f1 | sed s'/DP=//' | sed s'/INDEL//' | sed '/^$/d' > temp.txt mv temp.txt C3_M_fltVCF.vcf.DP.txt
Scenario 13
Have to seek for faster SNV calling with samtools, I found this –> mpileup function is very helpful
Solution
With xargs, I parallel the snp call by chromosomes (-P is the number of parallel processes started parallel
samtools view -H yourFile.bam | grep "\@SQ" | sed 's/^.*SN://g' | cut -f 1 | xargs -I {} -n 1 -P 24 sh -c "samtools mpileup -BQ0 -d 100000 -uf yourGenome.fa -r {} yourFile.bam | bcftools view -vcg - > tmp.{}.vcf"
Then merge the results afterwards
samtools view -H yourFile.bam | grep "\@SQ" | sed 's/^.*SN://g' | cut -f 1 | perl -ane 'system("cat tmp.$F[0].vcf >> yourFile.vcf");'
Scenario 14
I have a list of imprinted SNP-genes across mouse genomes, I want to know how many SNP/genes in each chromosome. Therefore, I need a bash loop script to quickly get an assessment.
Solution:
for ((i=1; i<=19; i++)); do echo chr$i; grep -w chr$i GSE27016_imprinted_genes.mm9.bed | wc -l ; echo; done ;
for i in {1..19}; do echo chr$i; done;
Scenario 15
I have Illumina exome capture region file and want to know how many bases are covered. It can be extended to ask for the sum of any column of numeric data.
Solution:
awk '{SUM +=$5} END {print SUM; }' TruSeq-Exome-65mb-BED-file_with_MT
62085295
awk '{SUM +=$3-$2} END {print SUM; }' SureSelect_All_Exon_37mb_hg19_bed_with_MT
38831632
awk '{SUM +=$3-$2} END {print SUM; }' SureSelect_All_Exon_50mb_hg19_bed_with_MT
51663197
Scenario 16
Du, Ying asks me to help her to append an extra unique ID column to a column file
awk '{print $1"_"$2"_"$3}' TruSeq-Exome-65mb-BED-file_with_MT > temp.txt
paste -d" " temp.txt TruSeq-Exome-65mb-BED-file_with_MT > temp2.txt
Scenario 17
In our FPKM file, I noticed duplicates “IDs”. What are those duplicate records?
cd ~/project2012/ASE/FPKM_RNAseq/FPKM
cut -f1 C3_M_3.mm9.genes.fpkm_tracking.fpkm.txt | perl -ne 'print if $SEEN{$_}++'
Scenario 18
In my ASE list, it has ^M at the end of each line. I need to remove them in Unix
To view it: cat -v ASE_gene_jyl.txt cat ASE_gene_jyl,txt | sed 's/^M//g' > temp mv temp ASE_gene_jyl.txt
Scenario 19
I am working on getting the structured snp list requested by Dr. Fargo, and I need to feed snp numbers with leading zero. The answer is not with unix script but a perl script
my $num = "123";
$num = sprintf("%06d", $num);
$num=~ tr/ /0/;
Scenario 20
Justin from NCSU needs help to modify his bed file for Galaxy. Input if like this:
1 0 3000236 0 1 3000236 3000272 1 1 3000272 3000311 0 1 3000311 3000347 1 1 3000347 3000628 0 1 3000628 3000645 1
And, he wanted:
chr1 0 3000236 0 chr1 3000236 3000272 1 chr1 3000272 3000311 0 chr1 3000311 3000347 1 chr1 3000347 3000628 0 chr1 3000628 3000645 1
Solution
awk '{print "chr"$1}' fileIN.txt > modCol1
cut -f2-4 fileIN.txt > reminders
paste modCol1 reminders > newFile
rm -f modCol1
rm -f reminders
Scenario 21: I wanted to extract file name from a string with path
../rawFastq/ES565_121204_MARMOSET_64NDWAAXX.2.3191261.single.sanger.fastq
Solution, well bash script
myString="../rawFastq/ES565_121204_MARMOSET_64NDWAAXX.2.3191261.single.sanger.fastq"
OIFS="$IFS"
IFS='/'
myArray=($myString)
IFS=$OIFS
echo ${#myArray[@]}
i=0
while [ $i -lt ${#myArray[@]} ]
do
echo "Element $i:${myArray[$i]}"
(( i=i+1 ))
done
#print out file name only
lastIndex=${#myArray[@]}-1
echo "${myArray[$lastIndex]}"
Scenario 22: I created unique 22mers from Rick’s microRNAseq project total 7257, and I wanted to get a fasta format of it
Solution, pure bash script
Head file looks like the following "x" "GAACCAGAAGGCTGTCTGAACT" "AGAAGGCTGTCTGAACTTTTGA" "AGGCTGTCTGAACTTTTGAAAC" "GGAACGAGTGTGAGTCTGAAAC" "GAACGAGTGTGAGTCTGAAACC" "AACGAGTGTGAGTCTGAAACCA" Step 1. Get rid of the first row: tail -n +2 overall_overlap.txt > temp.seq mv temp.seq overall_overlap.txt Step 2. Remove the "quotes": cat overall_overlap.txt | sed 's/\"//g' > temp.seq mv temp.seq overall_overlap.txt Step 3. Final script: for ((i=1; i\seq"$i; sed -n $i", "$i"p" overall_overlap.txt ; done > overall_overlap.fa
Scenario 23: There was a weird error with picard tool, and I need to delete a few lines in a large .sam file
It appears that it is unhappy to find two header lines for Bismark in the header (lines that start with "@PG ID:Bismark"). Very weird. If I take the first 1000 lines of your SAM file and put them in a new file, remove one of the "@PG ID: Bismark" lines, and run sort then it doesn't give an error. Interesting that I've ne ver seen this error when working with these files. Anyway, if you get rid of all but one of the @PG lines, it should be fine. That might be a pain. Sorry. I think Picard has a "ReplaceHeader" utility (not sure of the exact name). I've never used it, but these might be an ideal time to try it out!
The solution is: sed command as following
I love this sed help doc
Scenario 24
I wanted to use the regular expression in bash script, and further create processing script
I found a useful online help. So, I wrote my script like the following:
#!/usr/bin/bash
bam=$1
re="([^/]+).bam.sorted.bam"
if [[ $bam =~ $re ]]; then
root=${BASH_REMATCH[1]}
fi
bcf=$root.var.raw.bcf
vcf=$root.var.flt.vcf
echo "samtools mpileup -C50 -uf /ddn/gs1/home/li11/project2014/Copeland/reference/hs_mit_seq.fasta $bam | bcftools view -bvcg - > $bcf"
echo "bcftools view $bcf | ~/tools/samtools-0.1.18/bcftools/vcfutils.pl varFilter -D100 > $vcf"
Scenario 25
Removing unnecessary characters in a file
- It has been this case in the past and with “cat” all hidden
- characters are shown.
- To remove them, here is the post
- perl -i -pe ‘s/\r//g’ inputfile : remove unseen special characters
- perl -i -pe ‘s/[^\x20-\x7f]//g’ examplefile : remove ALL unseen
- special characters
- Sometimes, it affects database columns and it needs to be
cleaned to avoid query not getting back expected results.
- This works better:
- tr -cd ‘\11\12\40-\176’ PipeIN input PipeOUT myfile2
Okay, it does happen to me that the ctrl-M did NOT work.
For those of you saying this doesn’t work, I think I might have your answer. Please note this line in the article: “Type the following command (to get ^M type CTRL+V followed by CTRL+M):” You can’t just type the carat symbol and a capital M. You have to hit CTRL-V and then hit CTRL-M. I had quite a time figuring that out…. but once you do it right, it works great.
Scenario 26
Find a match and get the next line(s) or previous (lines)
Get the next line
- grep -A1 some-file will do it
- cat ../FastQC/MES1192_170504_M00421_B55HN.12-5224-1132-2.2.sanger_fastqc/fastqc_data.txt | grep -A2 “Overrepresented”
Get the previous line
- cat adapter_condition.txt | grep -B1 “>>Adapter Content”
Scenario 27 — different checksum method
There are at least three checksum methods that one can encounter
a file Mus_musculus.GRCm38.cds.all.fa.gz from Ensembl. They can be different results and cause confusion.
bash-4.2$ sum Mus_musculus.GRCm38.cds.all.fa.gz 40501 17135 bash-4.2$ cksum Mus_musculus.GRCm38.cds.all.fa.gz 414929315 17545827 Mus_musculus.GRCm38.cds.all.fa.gz bash-4.2$ md5sum Mus_musculus.GRCm38.cds.all.fa.gz 9af75c6fe212439e45c58357f4e1cdac Mus_musculus.GRCm38.cds.all.fa.gz
Scenario 28 — securely copy files in Linux
I have done some analysis on the server and want to securely transfer them onto my local linux machine. the command is “scp”, and an example would be something like the following:
scp li11@scigate.niehs.nih.gov:project2019/RNAseqProj/results/DE/DE_adjp05.zip .
Protected: Working with master project
Adding read group information
There has been much more headache adding this piece of information than I have expected. And, I think it is worthwhile to create a new thread just for this topic.
Ultimately, we would like to add this information into our SAMPE/SAMSE step using the -r option.
Working directory: /home/chavi/jl407/GATK-testing/testAddRG
Recalibration steps and expected output files:
Step 1 This should disappear once the new pipeline is updated (a -r option should be added at BWA step), for now, we need to manually add “read group” information and merge the bam files. But, there has been an error using this option .
Another source with details on the samtools options are here .
Merge BAM files with read group information, and further processing the bam file etc.: sampleName.RG.rmdup.bam
Just for testing purposes, will add read group into each individual bam file. A help was posted at SEQanswers forum by freeseek as:
echo -e “@RGtID:gatSM:hstLB:gatPL:Illumina” > rg.txt
samtools view -h ga.bam | cat rg.txt – | awk ‘{ if (substr($1,1,1)==”@”) print; else printf “%stRG:Z:gan”,$0; }’ | samtools view -uS – | samtools rmdup – – | samtools rmdup -s – aln.bam
With my modification as
echo -e “@RGtID:gatSM:hstLB:gatPL:Illumina” > rg.txt
samtools view -h ga.bam | cat rg.txt – | awk ‘{ if (substr($1,1,1)==”@”) print; else printf “%stRG:Z:gan”,$0; }’ | samtools view -bS -o aln.bam –
It did NOT work!!!
Take a look at the following screen shot:
[jl407@head4 testAddRG]$ samtools view -H /nfs/chgv/seqanalysis/ALIGNMENT/samples/als9c2/Run1/s.3.RG.bam
@RG ID:Run_1_3 SM:Run_1_3 LB:ga PL:Illumina
[jl407@head4 testAddRG]$ samtools view /nfs/chgv/seqanalysis/ALIGNMENT/samples/als9c2/Run1/s.3.RG.bam | more
…………….
XT:A:U NM:i:0 SM:i:37 AM:i:37 X0:i:1 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:100 RG:Z:ga
………………
Each step takes 20 minutes!!
To view where the “read group has been properly” added, use the samtools view option.
OK, let’s try the most dumb way!!!
Use script: /nfs/chgv/seqanalysis/ALIGNMENT/samples/als9c2/addRG.sh, which sends this command:
/nfs/chgv/seqanalysis/SOFTWARE/samtools-0.1.12a/samtools merge -rh /nfs/chgv/seqanalysis/ALIGNMENT/samples/als9c2/rg.txt – /nfs/chgv/seqanalysis/ALIGNMENT/samples/als9c2/Run1/s.2.bam /nfs/chgv/seqanalysis/ALIGNMENT/samples/als9c2/Run1/s.3.bam /nfs/chgv/seqanalysis/ALIGNMENT/samples/als9c2/Run1/s.4.bam /nfs/chgv/seqanalysis/ALIGNMENT/samples/als9c2/Run1/s.5.bam /nfs/chgv/seqanalysis/ALIGNMENT/samples/als9c2/Run2/s.7.bam /nfs/chgv/seqanalysis/ALIGNMENT/samples/als9c2/Run2/s.8.bam | samtools rmdup – – | samtools rmdup -s – als9c2_rmdup_RG.bam
Sent to run @ May 18th, 9:11 a.m. Well, it worked!! Finished @ May 19th, 23:04
However, I need to “sort” it at the last step.
Test-drive assessment on als9c2
Following the assessment thread done earlier , doing the real assessment on als9c2 with the focus on the following metrics.
1. ti/tv ratio
2. number of raw and filtered SNV
3. number of indel called by samtools
4. proportion of heterozygous SNVs on the x chr compared to the total number of SNVs in the X
5. concordance of SNVs with genotyping chip computed with SVA
6. overlap of filtered autosomal SNVs with dbsnp
7. Ratio of homo to hetero SNVs
For raw bam files
Here are the major steps
Input files: a bam file (with or without RG and/or recalibration)
Systems involved: dscr & lsrc
Software involved: samtools
Step One: get variant call file (samtools mpileup –> vcf format)
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b37.sh
2. Input: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/combined_rmdup.bam
3. OutputDir: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/
4. Submit job: qsub /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b37.sh /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/combined_rmdup.bam /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/(This has normally been done for raw bam file in the existing pipeline)
There has been error complaining about samtools mpileup, it turns out that it is error with read group information! Well, re-did the “add read group” step, still NOT working!!
samtools: bam_plcmd.c:596: group_smpl: Assertion `id >= 0 && id n’ failed.
On the recalibrated bam , no_RG case file (without RG due to error)
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b37.sh
2. Input: /nfs/chgv/seqanalysis2/GATK/recalBam/als9c2.build37.recal.noRG.bam
3. OutputDir: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/
4. Submit job: qsub /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b37.sh /nfs/chgv/seqanalysis2/GATK/recalBam/als9c2.build37.recal.noRG.bam /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/
5. Sent May 18th, 2011 @10:25 a.m.; finished by May 23rd @4:49 a.m.
Unfortunately, there has been errors with process, ended up in variant call truncated at chromosome 9! Could be “disk space issue”.
Comparing size of file1:
For no_RG case:
[jl407@head4 noRG_reCal]$ ls -al file1
-rw-r–r– 1 jl407 chavi 264284334703 May 23 04:49 file1
For raw bam case:
[jl407@head4 noRG_reCal]$ ls -al /nfs/chgv/seqanalysis2/ALIGNMENT/samples/als9c2/build37/combined/file1
-rw-r–r– 1 jl407 chavi 264495324652 May 4 08:52 /nfs/chgv/seqanalysis2/ALIGNMENT/samples/als9c2/build37/combined/file1
Compare bam file:
[jl407@head4 noRG_reCal]$ ls -al /nfs/chgv/seqanalysis2/GATK/recalBam/
total 157200748
drwxr-xr-x 2 jl407 chavi 4096 May 18 11:39 .
drwxr-xr-x 11 jl407 chavi 4096 May 23 15:11 ..
-rw-r–r– 1 jl407 chavi 160649148678 May 18 10:13 als9c2.build37.recal.noRG.bam
-rw-r–r– 1 jl407 chavi 8763904 May 18 11:39 als9c2.build37.recal.noRG.bam.bai
[jl407@head4 noRG_reCal]$ ls -al /nfs/chgv/seqanalysis2/ALIGNMENT/samples/als9c2/build37/combined/combined_rmdup.bam
-rw-r–r– 1 jl407 chavi 98071917844 Apr 29 14:23 /nfs/chgv/seqanalysis2/ALIGNMENT/samples/als9c2/build37/combined/combined_rmdup.bam
Need to re-run this, starts on May 24th, @ 4:00 p.m.
Step two: Split vcf files by snps and indels
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl
2. Input: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/var_flt_vcf
3. RunningDir/OutputDir: /nfs/chgv/seqanalysis2/GATK/runningDir
4. cd to the RunningDir, and submit job:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl /nfs/chgv/seqanalysis2/GATK/runningDir/genome_vcf_files, this file contains a list of var_flt_vcf to process.als9c2 /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/var_flt_vcf
For no_RG case:
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl
2. Input: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/var_flt_vcf
3. RunningDir/OutputDir: /nfs/chgv/seqanalysis2/GATK/runningDir
4. cd to the RunningDir, and submit job:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl /nfs/chgv/seqanalysis2/GATK/runningDir/genome_vcf_files, this file contains a list of var_flt_vcf to process.als9c2 /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/var_flt_vcf
Step three: Filter vcf variants
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl
2. Input: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/var_flt_vcf.snp
3. RunningDir/OutputDir: /nfs/chgv/seqanalysis2/GATK/runningDir
4. cd to the RunningDir, and submit job:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl /nfs/chgv/seqanalysis2/GATK/runningDir/vcf_snp_filelist snpvcf_snp_filelist contains a list of var_flt_vcf.snp to process.
als9c2 /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/var_flt_vcf.snp
For no_RG case:
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl
2. Input: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/var_flt_vcf.snp
3. RunningDir/OutputDir: /nfs/chgv/seqanalysis2/GATK/runningDir
4. cd to the RunningDir, and submit job:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl /nfs/chgv/seqanalysis2/GATK/runningDir/vcf_snp_filelist snpvcf_snp_filelist contains a list of var_flt_vcf.snp to process.
als9c2 /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/var_flt_vcf.snp
Result: should be in the following format, two files with the same name as your variant file will be created:
.filtered (variants that passed QC threshold)
.filtered.out (variants which failed QC)
Step four: ti/tv ratio
Dependency:
Java path: export PATH=/home/csem/tm103/DSCR/jre1.6.0_13/bin:$PATH
GATK package: gatk_dir=/nfs/chgv/seqanalysis/SOFTWARE/GenomeAnalysisTK-1.0.5083
Reference DB (build 37): reference=/nfs/chgv/seqanalysis/ALIGNMENT/BWA_INDEX_JYL/human_g1k_v37.fasta
Commands to issue
1. Samples ( als9c2) dir:/nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/
2. Script :/nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv.sh
3. Log files: /nfs/chgv/seqanalysis2/GATK/logs/
4. Output: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/gatk_titv.csv
5. Command: qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv.sh /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/
For no_RG case: — Changes in Italic
1. Two input information:
a. Samples ( als9c2) dir:/nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/
b. Output file name: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/als9c2.build37.noRG.recal.gatk_titv.csv
2. Script :/nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv.sh
3. Log files: /nfs/chgv/seqanalysis2/GATK/logs/
4. Command: qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv.sh /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/ als9c2.build37.noRG.recal.gatk_titv.csv
FIXME: this output needs to be “sample” oriented!!!, i.e. als9c2.build37.precal.gatk_titv.csv
Step five: Homozygous/heterozygous SNV ratio
Check to see if the genotype is listed in the 10th column in the VCF file
Script: /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl
Usage: perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl vcf_file > printTO
Procedure: cd to “/nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/” and issue command: perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/var_flt_vcf.snp.filtered > als9c2.build37.precal.homhet.snp.ratio.hets
For noRG_recal case:
Script: /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl
Usage: perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl vcf_file > printTO
Procedure: cd to “/nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/” and issue command: perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/var_flt_vcf.snp.filtered > als9c2.build37.noRG.recal.homhet.snp.ratio.hets
Step six: Concordance check with SVA
(1) After the “variant call” in step one, generate bco files in the DSCR
Use the following 2 lines from this script
/nfs/chgv/seqanalysis2/GATK/scripts/bcftools_coverage_bco.sh:
Modify all the parameters, including Java path, samtools location, reference DB, etc.
export PATH=/home/csem/tm103/DSCR/jre1.6.0_13/bin:$PATH
sam_dir=/nfs/chgv/seqanalysis/SOFTWARE/samtools-0.1.12a
reference_fasta_file=/nfs/chgv/seqanalysis/ALIGNMENT/BWA_INDEX_JYL/human_g1k_v37.fasta
If it runs manually, a few subdirectories need to be created properly.
a. bco_dir=$combined_dir/bco
b. $bco_dir/$sample
c. seqsata_sample_dir=/nfs/testrun/GATK-recal/assessmentComp/samples/$sample (for the results to be transferred back to our sva clusters).
Example command:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/bcftools_coverage_bco_build37.sh /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/ /nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal
/nfs/chgv/seqanalysis/SOFTWARE/samtools-0.1.12a/bcftools/bcftools view -cg /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal//file1 > /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal//bcftools_file
java -jar /nfs/chgv/seqanalysis2/GATK/scripts/vcf2bco.jar /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal//bcftools_file /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal//bco/
rsync -a –partial –timeout=10000 -e ‘ssh’ -r /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal//bco sva6.lsrc.duke.edu:/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal
For noRG_case:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/bcftools_coverage_bco_build37.sh /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/ /nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/noRG_reCal
/nfs/chgv/seqanalysis/SOFTWARE/samtools-0.1.12a/bcftools/bcftools view -cg /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal//file1 > /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal//bcftools_file
java -jar /nfs/chgv/seqanalysis2/GATK/scripts/vcf2bco.jar /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal//bcftools_file /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal//bco/
rsync -a –partial –timeout=10000 -e ‘ssh’ -r /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal//bco sva6.lsrc.duke.edu:/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/noRG_reCal
At last step, those bco files should be transferred back to LSRC
(2) Create an SVA project (meaning get a .gsap file created)
Since this is alignment against build 37, there are many changes to the sva project. Thanks to Dongliang, who helped me with the set ups:
Currently, The new build is existing at: /nfs/goldstein/goldsteinlab/software/SequenceVariantAnalyzer_1.1_b37/
The .gsap file needs to be modified accordingly:
1. Menu File -> Open example project file
2. Copy all the lines after line “#Reference database version”
3. Replace the lines in the same place in your build 36 .gsap project file 4. Remove all the starting “#” in the “[REFBIN]=” lines. Because your project has all chromosomes, while the example only has X.
4. At last, Dongliang had to modified the permission for ./datasource/ensembl/61_37f/variation_feature2.txt
MODIFIED .gsp file!! May 18th, 2011
.GSAP file for SVA project: /nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal/vcf_als9c2_precal.gsap
(3) Topstrand file names for als9c2:
/nfs/testrun/GATK-recal/svaprojects/vcf/genome/topstrandfilenames (Illumina created/provided this file)
/nfs/testrun/GATK-recal/svaprojects/vcf/genome/topstrandfilenames, the content in this file is as following.
SAMPLE_ID GENOTYPING_ID TopStrand_files
als9c2, als009c2, /nfs/sva01/SequenceAnalyses/TopStrandFiles/als006-015_10samples__MAY_4_10_TopStrand.csv
(4) Use this version of SVA: (It is about 120 mitues.
/nfs/goldstein/goldsteinlab/software/SequenceVariantAnalyzer_1.1_b37/
(Make sure to uncheck the filtering option in sva)
With Xwin, I was able to ssh to sva0.lsrc.duke.edu and perform the “annotation” . Started @7:30 a.m. May 19th, 2011.
(5) Check concordance:
SVA version: /nfs/goldstein/goldsteinlab/software/SequenceVariantAnalyzer_1.1_b37/
Case I: Once “annotate” is done, go directly to the following steps to get concordance.
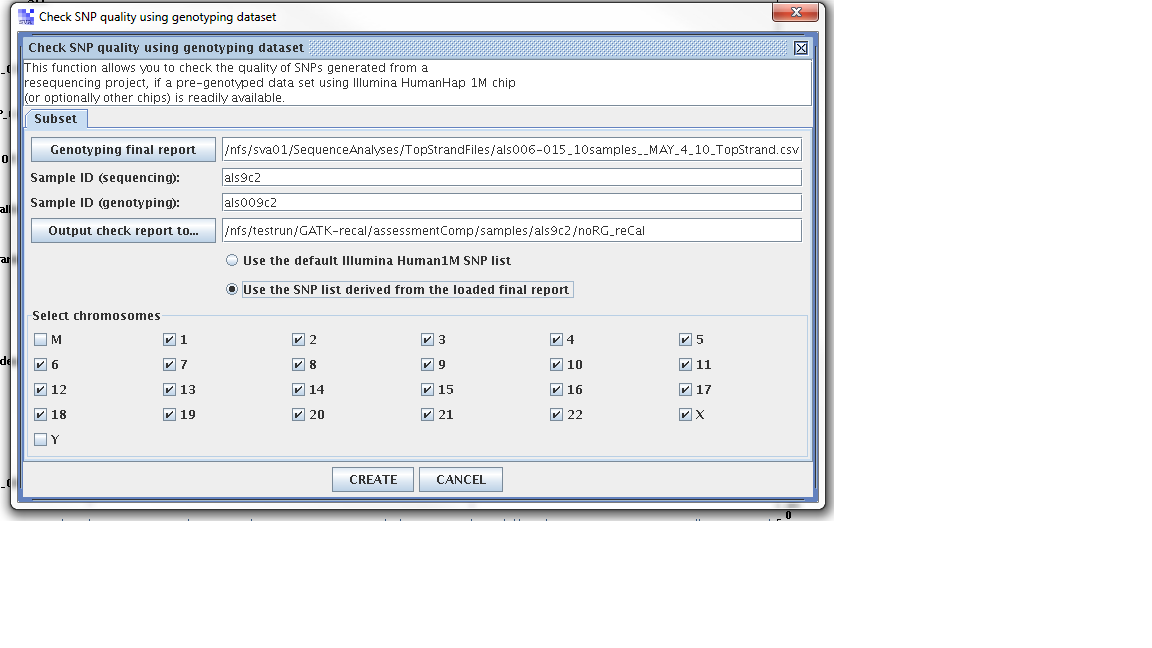
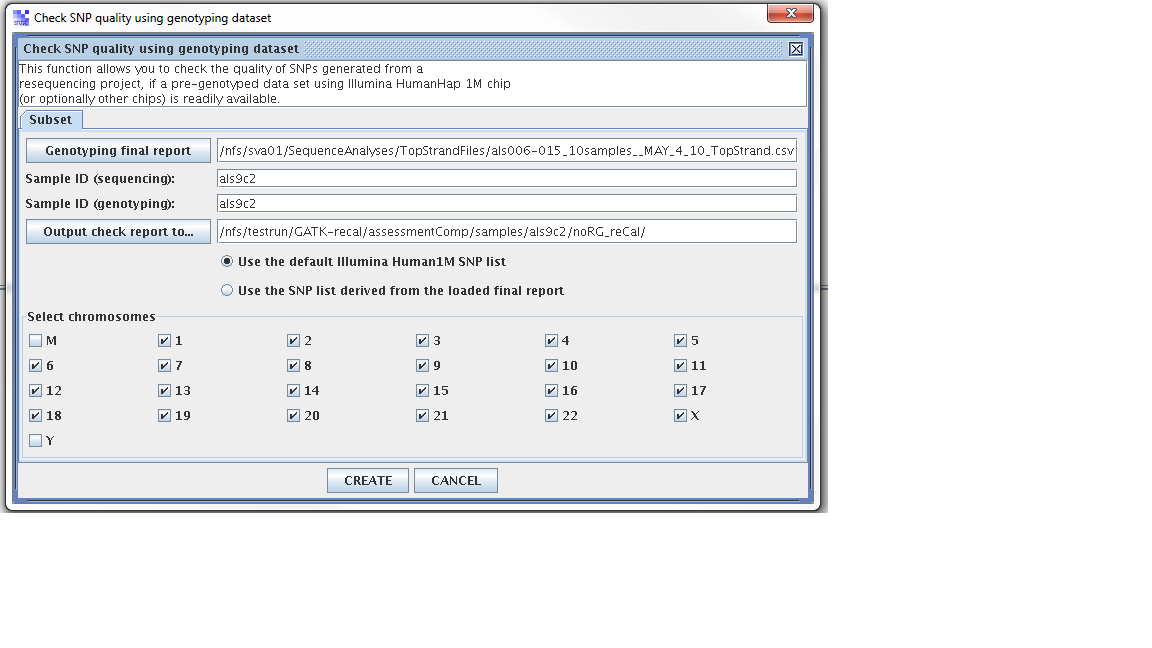
Step 1. From the SVA UI, choose “summary -> check SNP quality using genotyping dataset”
Step 2: Filling in information for four fields, information is stored at
Four fields
Genoptying final report: //nfs/sva01/SequenceAnalyses/TopStrandFiles/als006-015_10samples__MAY_4_10_TopStrand.csv
sample ID (sequencing): als9c2
sample ID (genotyping): als009c2
output check report to: /nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal
Step 3: Check “use the SNP list derived from the loaded final report”
Step 4: Click “Create”
Case II: If the project is closed, load the project by clicking “load” button and go back to the steps mentioned above.
Results:
For pre-calibration case
Performing quality check using genotype data (user SNP list).
> Sample ID (sequencing): als9c2
Sample ID (genotyping): als009c2
Note: 584882 SNPs are loaded from user SNP report.
– Chromosome: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X
– 297962 SNPs (with rs# listed in genotype data (user SNP list) are loaded from your resequencing project.
– 148881 SNPs on bottom strand (Illumina).
– Among these, 296597 SNPs are found in genotype data (user SNP list) .
– Among the 297962 SNPs loaded from resequencing project, there are:
294755 match with genotype data (user SNP list);
1842 mismatch;
1365 not found in genotype data (user SNP list).
– Among the 1842 mismatches, there are
21 hom (seq) – hom (geno)
32 hom (seq) – het (geno)
18 het (seq) – het (geno)
1771 het (seq) – hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal.nomatch.txt
[Part 2] N = 286770
– Among the 286770 SNPs included in the genotyping chip but not derived from sequencing, there are:
286767 that can be correctly mapped to reference sequence;
Among these 286767 there are 281402 matches;
and 5365 mismatches, including 3699 het (geno) and 1666 hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal.part2.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal.part2.nomatch.txt
Results for recalibrated results
Performing quality check using genotype data (user SNP list).
> Sample ID (sequencing): als9c2
Sample ID (genotyping): als009c2
Note: 584882 SNPs are loaded from user SNP report.
– Chromosome: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X
– 299193 SNPs (with rs# listed in genotype data (user SNP list) are loaded from your resequencing project.
– 149493 SNPs on bottom strand (Illumina).
– Among these, 297794 SNPs are found in genotype data (user SNP list) .
– Among the 299193 SNPs loaded from resequencing project, there are:
296497 match with genotype data (user SNP list);
1297 mismatch;
1399 not found in genotype data (user SNP list).
– Among the 1297 mismatches, there are
21 hom (seq) – hom (geno)
32 hom (seq) – het (geno)
20 het (seq) – het (geno)
1224 het (seq) – hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/noRG_reCal.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/noRG_reCal.nomatch.txt
[Part 2] N = 285979
– Among the 285979 SNPs included in the genotyping chip but not derived from sequencing, there are:
282538 that can be correctly mapped to reference sequence;
Among these 282538 there are 138818 matches;
and 143720 mismatches, including 2429 het (geno) and 141291 hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/noRG_reCal.part2.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/noRG_reCal.part2.nomatch.txt
As the results show, we see improvement on the concordance with both part I and part II. And, it is more important that we see improvement with part II.
So the question exists, why/what causes low concordance check with genotyp chip results???
Step seven: Overlap of filtered autosomal SNVs with dbsnp
Here we need a little bit more attention. Since we are trying to compare analysis against build 37, we need snp information for build 37. It is available at NCBI/dbsnp
Download dbsnp file as following:
1. Download b132_SNPChrPosOnRef_37_1.bcp.gz
2. Check download completeness
[jl407@head4 refDB]$ md5sum b132_SNPChrPosOnRef_37_1.bcp.gz
07acf58908cf857b62e9de99a1d56533 b132_SNPChrPosOnRef_37_1.bcp.gz
[jl407@head4 refDB]$ cat b132_SNPChrPosOnRef_37_1.bcp.gz.md5
MD5(b132_SNPChrPosOnRef_37_1.bcp.gz)= 07acf58908cf857b62e9de99a1d565331. Samples (info) located: /nfs/chgv/seqanalysis2/GATK/doc/snplistFile
2. Script (dbsnp_overlap.pl) located: /nfs/chgv/seqanalysis2/GATK/scripts/
3. Log files: /nfs/chgv/seqanalysis2/GATK/logs/
4. Command: qsub /nfs/chgv/seqanalysis2/GATK/scripts/dbsnp_overlap_b37.pl /nfs/chgv/seqanalysis2/GATK/doc/snplistFile
5. Results were written to log file
For no_RG case:
The only change is where the var_flt_vcf.snp.filtered is located:
als9c2 /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/var_flt_vcf.snp.filtered
als9c2 /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/var_flt_vcf.snp.filtered
Here is the concordance checking results: concordance_als9c2_b37
July 19th, 2011
Wanted to test “using 1M vs snp loaded from the project”.
Test run on NA12878
Refer to the page on work@chgv
Without ReadGroup
There has been an error with adding the readgroup information, therefore, I am testing this procedure just like I did with earlier trial (without read group information)
Getting ready for NA12878 sampel recalibration:
Step 1: making symbolic links to raw bam file and raw bai file
Step 2: Send this script and start the process raw bam –> cntcor.csv –> recal bam –> index recal bam –> recal.cntcor.csv
Step 3: send the following command:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/GATK_recal.pl na12878 /nfs/chgv/seqanalysis2/GATK/rawBam/na12878.build37.raw.bam /nfs/chgv/seqanalysis2/GATK/resultsDir/na12878.build37.raw.bam.noRG.CntCov.csv /nfs/chgv/seqanalysis2/GATK/recalBam/na12878.build37.recal.noRG.bam /nfs/chgv/seqanalysis2/GATK/resultsDir/na12878.build37.recal.bam.noRG.CntCov.csv
java -jar -Xmx12g /nfs/chgv/seqanalysis/SOFTWARE/GenomeAnalysisTK-1.0.5083//GenomeAnalysisTK.jar
-T CountCovariates
-nt 12
-R /nfs/chgv/seqanalysis/ALIGNMENT/BWA_INDEX_JYL/human_g1k_v37.fasta
-B:eval,VCF /home/chavi/jl407/GATK-testing/referenceFiles/00-All.vcf
-I /nfs/chgv/seqanalysis2/GATK/rawBam/na12878.build37.raw.bam
–default_read_group na12878
–default_platform illumina
-cov ReadGroupCovariate
-cov QualityScoreCovariate
-cov DinucCovariate
-cov CycleCovariate
-recalFile /nfs/chgv/seqanalysis2/GATK/resultsDir/na12878.build37.raw.bam.noRG.CntCov.csv
java -jar -Xmx12g /nfs/chgv/seqanalysis/SOFTWARE/GenomeAnalysisTK-1.0.5083//GenomeAnalysisTK.jar
-T TableRecalibration
-R /nfs/chgv/seqanalysis/ALIGNMENT/BWA_INDEX_JYL/human_g1k_v37.fasta
-I /nfs/chgv/seqanalysis2/GATK/rawBam/na12878.build37.raw.bam
–default_read_group na12878
–default_platform illumina
-recalFile /nfs/chgv/seqanalysis2/GATK/resultsDir/na12878.build37.raw.bam.noRG.CntCov.csv
–out /nfs/chgv/seqanalysis2/GATK/recalBam/na12878.build37.recal.noRG.bam
/nfs/chgv/seqanalysis/SOFTWARE/samtools-0.1.12a/samtools index /nfs/chgv/seqanalysis2/GATK/recalBam/na12878.build37.recal.noRG.bam
java -jar -Xmx12g /nfs/chgv/seqanalysis/SOFTWARE/GenomeAnalysisTK-1.0.5083//GenomeAnalysisTK.jar
-T CountCovariates
-nt 12
-R /nfs/chgv/seqanalysis/ALIGNMENT/BWA_INDEX_JYL/human_g1k_v37.fasta
-B:eval,VCF /home/chavi/jl407/GATK-testing/referenceFiles/00-All.vcf
-I /nfs/chgv/seqanalysis2/GATK/recalBam/na12878.build37.recal.noRG.bam
–default_read_group na12878
–default_platform illumina
-cov ReadGroupCovariate
-cov QualityScoreCovariate
-cov DinucCovariate
-cov CycleCovariate
-recalFile /nfs/chgv/seqanalysis2/GATK/resultsDir/na12878.build37.recal.bam.noRG.CntCov.csv
Help needed from Jessica
Since Jessica has compared 1000 genome sample obtained locally to snp released from 1000 Genome sites, I would like to learn what she had gone through. The following would get me started:
1. Where is snp file stored locally
2. Assuming we have gone through the entire process, we should have:
a. ram bam (aligned to build36) with index
b. output from call_variant script using “samtools12”, maybe a copy from “samtools7”
3. Since Jessica does not have a way to compare vcf format directly, she converted VCF format SNP back to pileup format
a. a script that does this conversion
b. where are the output from the conversion
4. Assuming Jessica has converted “1000 genome vcf snp” to “pileup” format
a. where are the pileup format 1000 genome snp file
5. Comparing script, that compares two pileup snp files
Jessica’s response
Hi Jianying,
These are the scripts I used for SNV comparison. I don’t have a routine that compares two VCF files and my conversion from VCF to pileup is not perfect.
This script converts a VCF SNV file to pileup format:
/nfs/chgv/seqanalysis/ALIGNMENT/QC/Scripts/evaluation/vcf_topileup_snps.pl
The genotype column in the VCF file is hard coded as 9 (see 5th line in the code). You can change this accordingly.Comparing two SNV files in pileup format:
I used this .sh script to do the SNP file comparison /nfs/chgv/seqanalysis/ALIGNMENT/QC/Scripts/evaluation/snp_file_comparison.sh
This is the perl script that is called inside the .sh script:
/nfs/chgv/seqanalysis/ALIGNMENT/QC/Scripts/evaluation/compare_snp_files.plJessica
Another email:
Hi Jianying,
Here are the answers to your questions:
1. Where is 1000 genome snp file stored locally? If we LOST it,
which file we should re-download from 1000 genome?
I think this file might have been deleted soon after Sam Dickson left.
I would recommend re-downloading it.2. Assuming we have gone through the entire process, we should have:
a. Raw bam (aligned to build36) with index and c. Output files from “samtools7″ (in pileup format) This is located here: /nfs/seqsata01/ALIGNMENT/BWA_OUTPUT/na12878/combinedb. Output files from call_variant script using “samtools12″ (in vcf format) /nfs/chgv/seqanalysis/ALIGNMENT/QC/samtools_version/sam12/genome/na12878/var_flt_vcf
3. Since we do not have a way to compare vcf format directly,
Jessica converted VCF format SNP back to pileup format a. The script that does this conversion Sent in my previous email this morning.4. Assuming Jessica has converted “1000 genome vcf snp” to “pileup” format
a. Where are the pileup format 1000 genome snp file Please use my script to do re-convert the VCF into pileup format.5. Comparing script, that compares two pileup snp files, and
report that it generated.
Sent in my previous email this morning.Please let me know if you have any additional questions.
Jessica
Help from Dongliang to download 1000 genome snp file in vcf format:
Here are the links to the download:
I downloaded: CEU.trio.2010_03.genotypes.vcf.gz to /nfs/testrun/GATK-recal/snp_1000G_b36
Here are the first 20 lines:
[jl407@sva snp_1000G_b36]$ head -20 CEU.trio.2010_03.genotypes.vcf
##fileformat=VCFv4.0
##INFO=
##INFO=
##INFO=
##INFO=
##reference=human_b36_both.fasta
##FORMAT=
##FORMAT=
##FORMAT=
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT NA12891 NA12892 NA12878
1 52066 rs28402963 T C . PASS AA=C;DP=84 GT:GQ:DP 1/0:44:23 1/0:43:20 1/0:70:36
1 695745 . G A . PASS AA=.;DP=124 GT:GQ:DP 1|0:100:34 0|0:62:20 1|0:100:56
1 742429 rs3094315 G A . PASS AA=g;DP=132;HM2 GT:GQ:DP 1|1:100:38 1|1:59:30 1|1:100:44
1 742584 rs3131972 A G . PASS AA=a;DP=160;HM3 GT:GQ:DP 1|1:100:50 1|1:100:33 1|1:100:60
1 744366 rs3115859 G A . PASS AA=g;DP=127 GT:GQ:DP 1|1:80:31 1|1:100:34 1|1:100:45
1 746243 rs3131963 T A . PASS AA=t;DP=105 GT:GQ:DP 1|1:52:29 1|1:43:24 1|1:100:56
1 746775 rs6699990 A G . PASS AA=N;DP=120 GT:GQ:DP 0|1:100:34 0|0:89:30 0|0:100:46
1 747503 rs3115853 G A . PASS AA=-;DP=113 GT:GQ:DP 1|1:100:37 1|1:61:25 1|1:100:27
1 747597 rs4951929 C T . PASS AA=c;DP=129 GT:GQ:DP 1|1:100:36 1|1:86:28 1|1:100:56
1 747799 rs4951862 C A . PASS AA=c;DP=97 GT:GQ:DP 1|1:67:26 1|1:44:13 1|1:100:62
Strategy:
1. Run “comparing script” on two pileup format snp file
2. Run “conversion script” on a given vcf format to pileup
3. Maybe download vcf format snp file from 1000 genome
4. Run “recal” on build 36 na12878 bam file -> calibrated bam -> variant
Test-drive recalibration on als9c2
Refer to the page on work@chgv
Without ReadGroup
There has been an error with adding the readgroup information, therefore, I am testing this procedure just like I did with earlier trial (without read group information)
Input file preparation
Step 1: CountCovariates:
Script: /nfs/chgv/seqanalysis2/GATK/scripts/gatk_CountCov_noRG.sh
sample: als9c2
inputBAM: /nfs/chgv/seqanalysis2/GATK/rawBam/als9c2.build37.raw.bam
output: /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.raw.bam.noRG.CntCov.csv
Command sent:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_CountCov_noRG.sh als9c2 /nfs/chgv/seqanalysis2/GATK/rawBam/als9c2.build37.raw.bam /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.raw.bam.noRG.CntCov.csv
java -jar -Xmx12g /nfs/chgv/seqanalysis/SOFTWARE/GenomeAnalysisTK-1.0.5083/GenomeAnalysisTK.jar
-T CountCovariates
-nt 12
-R /nfs/chgv/seqanalysis/ALIGNMENT/BWA_INDEX_JYL/human_g1k_v37.fasta
-B:eval,VCF /home/chavi/jl407/GATK-testing/referenceFiles/00-All.vcf
-I /nfs/chgv/seqanalysis2/GATK/rawBam/als9c2.build37.raw.bam
–default_read_group als9c2
–default_platform illumina
-cov ReadGroupCovariate
-cov QualityScoreCovariate
-cov DinucCovariate
-cov CycleCovariate
-recalFile : /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.raw.bam.noRG.CntCov.csv
2. Step two: TableCalibration:
Script: /nfs/chgv/seqanalysis2/GATK/scripts/gatk_TableRecal_noRG.sh
sample: als9c2
inputBAM: /nfs/chgv/seqanalysis2/GATK/rawBam/als9c2.build37.raw.bam
tabRecalFile: /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.raw.bam.noRG.CntCov.csv
recalBAM: /nfs/chgv/seqanalysis2/GATK/recalBam/als9c2.build37.recal.noRG.bam
Command sent:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_TableRecal_noRG.sh als9c2 /nfs/chgv/seqanalysis2/GATK/rawBam/als9c2.build37.raw.bam /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.raw.bam.noRG.CntCov.csv /nfs/chgv/seqanalysis2/GATK/recalBam/als9c2.build37.recal.noRG.bam
java -jar -Xmx12g GenomeAnalysisTK.jar
-T TableRecalibration
-R /nfs/chgv/seqanalysis/ALIGNMENT/BWA_INDEX_JYL/human_g1k_v37.fasta
-I /nfs/chgv/seqanalysis2/GATK/rawBam/als9c2.build37.raw.bam
–default_read_group als9c2
–default_platform illumina
-recalFile /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.raw.bam.noRG.CntCov.csv
–out /nfs/chgv/seqanalysis2/GATK/recalBam/als9c2.build37.recal.noRG.bam
*Note,
-outputBam is deprecated, use -–out instead
Currently, not supporting parallel, only single thread
Time: 22.75 hours
2.1. Step 2.1: Indexing recalibrated BAM file:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_indexBAM.sh /nfs/chgv/seqanalysis2/GATK/recalBam/als9c2.build37.recal.noRG.bam
3. Step three: CountCovariates (on recalibrated bam file):
Script: /nfs/chgv/seqanalysis2/GATK/scripts/gatk_CountCov_noRG.sh
sample: als9c2
inputBAM: /nfs/chgv/seqanalysis2/GATK/recalBam
/als9c2.build37.recal.noRG.bam
output: /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.recal.bam.noRG.CntCov.csv
Command sent:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_CountCov_noRG.sh als9c2 /nfs/chgv/seqanalysis2/GATK/recalBam/als9c2.build37.recal.noRG.bam /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.recal.bam.noRG.CntCov.csv
java -jar -Xmx12g /nfs/chgv/seqanalysis/SOFTWARE/GenomeAnalysisTK-1.0.5083/GenomeAnalysisTK.jar
-T CountCovariates
-nt 12
-R /nfs/chgv/seqanalysis/ALIGNMENT/BWA_INDEX_JYL/human_g1k_v37.fasta
-B:eval,VCF /home/chavi/jl407/GATK-testing/referenceFiles/00-All.vcf
-I /nfs/chgv/seqanalysis2/GATK/recalBam/als9c2.build37.recal.noRG.bam
–default_read_group als9c2
–default_platform illumina
-cov ReadGroupCovariate
-cov QualityScoreCovariate
-cov DinucCovariate
-cov CycleCovariate
-recalFile /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.recal.bam.noRG.CntCov.csvTotally: 8.9 hours
4. Step four: AnalyzeCovariates (on raw bam file):
java -jar -Xmx6g $gatk_dir/AnalyzeCovariates.jar
-outputDir $outputDir
-resources $resourcesDir
-Rscript $RDir (/opt/R271/bin/R) , we have to provide this Rscript parameter.
-recalFile $tabRecalFile
Command sent: qsub /scriptdir/gatk_analCov.sh /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.raw.bam.noRG.CntCov.csv /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/
FIXME: BUT, the problem is that R script did not generate corresponding figures.
5. Step five: AnalyzeCovariates (on recalibrated bam file):
java -jar -Xmx6g $gatk_dir/AnalyzeCovariates.jar
-outputDir $outputDir
-resources $resourcesDir
-Rscript $RDir (/opt/R271/bin/R) , we have to provide this Rscript parameter.
-recalFile $tabRecalFile
Command sent: qsub /scriptdir/gatk_analCov.sh /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.recal.bam.noRG.CntCov.csv /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/
!! Need to fix the R code for assessment plots
With ReadGroup
ERRORS
When used samtools mpileup, I found this error “[mpileup] 1 samples in 1 input files
samtools: bam_plcmd.c:596: group_smpl: Assertion `id >= 0 && id n’ failed.” and later found out that the RG was not added as I expected. Here are the answer to my question at SEQanswers. Therefore, I have to recreate all the recalibrated bam files.
Use the dumb way to add ReadGroup information, hopefully it works. Fingers crossed
If it works, a pipeline GATK run will be fired up after checking the bam file and indexing it.
Step one: Check he bam file
samtools view -H /nfs/chgv/seqanalysis/ALIGNMENT/samples/als9c2/als9c2_rmdup_RG.bam
[jl407@head4 als9c2]$ samtools view -H als9c2_rmdup_RG.bam
@RG ID:Run_1_2 SM:Run_1_2 LB:ga PL:Illumina
@RG ID:Run_1_3 SM:Run_1_3 LB:ga PL:Illumina
@RG ID:Run_1_4 SM:Run_1_4 LB:ga PL:Illumina
@RG ID:Run_1_5 SM:Run_1_5 LB:ga PL:Illumina
@RG ID:Run_2_7 SM:Run_2_7 LB:ga PL:Illumina
@RG ID:Run_2_8 SM:Run_2_8 LB:ga PL:Illumina
samtools view /nfs/chgv/seqanalysis/ALIGNMENT/samples/als9c2/als9c2_rmdup_RG.bam | grep “RG”
HWI-ST426_0113:3:1101:1497:1989#0 99 1 177398310 29 100M = 177398565 355 ATTTGTCCAAAATGAGGAATACAATCTCTTATTCCAAAGAGCTATAAAAAAGAAGATAATCAATCTACT
TGAGAAGCCAATTGTTGGCATACATAAAGGC 6-./11142D<@DD@D;DD@6@@@??9??;?=7?<:6::99942555535=;=6<66<<?6<?=::6:7<9<DD=9=??<==6496145<494 XT:A:U NM:i:1 SM:i:29 AM:i:29 X0:i:1 X1:i:0 XM:i:
1 XO:i:0 XG:i:0 MD:Z:92C7 RG:Z:s.3
Step two: mv the bam file to /nfs/chgv/seqanalysis2/GATK/rawBam/
Step three: index the bam file
qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_indexBAM.sh /nfs/chgv/seqanalysis2/GATK/rawBam/
Step four: fire up the “GATK pipeline”
qsub /nfs/chgv/seqanalysis2/GATK/scripts/GATK_recal_RG.pl als9c2 /nfs/chgv/seqanalysis2/GATK/rawBam/als9c2_rmdup_RG.bam /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.raw.bam.RG.CntCov.csv /nfs/chgv/seqanalysis2/GATK/recalBam/als9c2.build37.recal.RG.bam /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.recal.bam.RG.CntCov.csv
Log file:
Process command:
java -jar -Xmx12g /nfs/chgv/seqanalysis/SOFTWARE/GenomeAnalysisTK-1.0.5083/GenomeAnalysisTK.jar
-T CountCovariates
-nt 12
-R /nfs/chgv/seqanalysis/ALIGNMENT/BWA_INDEX_JYL/human_g1k_v37.fasta
-B:eval,VCF /home/chavi/jl407/GATK-testing/referenceFiles/00-All.vcf
-I /nfs/chgv/seqanalysis2/GATK/rawBam/als9c2_rmdup_RG.bam
-cov ReadGroupCovariate
-cov QualityScoreCovariate
-cov DinucCovariate
-cov CycleCovariate
-recalFile /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.raw.bam.RG.CntCov.csvTime to start the process is: Thu May 19 11:37:59 2011
Time when the process finishes is: Thu May 19 11:37:59 2011Process command:
java -jar -Xmx12g /nfs/chgv/seqanalysis/SOFTWARE/GenomeAnalysisTK-1.0.5083/GenomeAnalysisTK.jar
-T TableRecalibration
-R /nfs/chgv/seqanalysis/ALIGNMENT/BWA_INDEX_JYL/human_g1k_v37.fasta
-I /nfs/chgv/seqanalysis2/GATK/rawBam/als9c2_rmdup_RG.bam
-recalFile /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.raw.bam.RG.CntCov.csv
–out /nfs/chgv/seqanalysis2/GATK/recalBam/als9c2.build37.recal.RG.bamTime to start the process is: Thu May 19 11:37:59 2011
Time when the process finishes is: Thu May 19 11:37:59 2011Process command:
/nfs/chgv/seqanalysis/SOFTWARE/samtools-0.1.12a/samtools index /nfs/chgv/seqanalysis2/GATK/recalBam/als9c2.build37.recal.RG.bamTime to start the process is: Thu May 19 11:37:59 2011
Time when the process finishes is: Thu May 19 11:37:59 2011Process command:
java -jar -Xmx12g /nfs/chgv/seqanalysis/SOFTWARE/GenomeAnalysisTK-1.0.5083/GenomeAnalysisTK.jar
-T CountCovariates
-nt 12
-R /nfs/chgv/seqanalysis/ALIGNMENT/BWA_INDEX_JYL/human_g1k_v37.fasta
-B:eval,VCF /home/chavi/jl407/GATK-testing/referenceFiles/00-All.vcf
-I /nfs/chgv/seqanalysis2/GATK/recalBam/als9c2.build37.recal.RG.bam
-cov ReadGroupCovariate
-cov QualityScoreCovariate
-cov DinucCovariate
-cov CycleCovariate
-recalFile /nfs/chgv/seqanalysis2/GATK/resultsDir/als9c2.build37.recal.bam.RG.CntCov.csvTime to start the process is: Thu May 19 11:37:59 2011
Time when the process finishes is: Thu May 19 11:37:59 2011