Following the assessment thread done earlier , doing the real assessment on als9c2 with the focus on the following metrics.
1. ti/tv ratio
2. number of raw and filtered SNV
3. number of indel called by samtools
4. proportion of heterozygous SNVs on the x chr compared to the total number of SNVs in the X
5. concordance of SNVs with genotyping chip computed with SVA
6. overlap of filtered autosomal SNVs with dbsnp
7. Ratio of homo to hetero SNVs
For raw bam files
Here are the major steps
Input files: a bam file (with or without RG and/or recalibration)
Systems involved: dscr & lsrc
Software involved: samtools
Step One: get variant call file (samtools mpileup –> vcf format)
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b37.sh
2. Input: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/combined_rmdup.bam
3. OutputDir: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/
4. Submit job: qsub /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b37.sh /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/combined_rmdup.bam /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/
(This has normally been done for raw bam file in the existing pipeline)
There has been error complaining about samtools mpileup, it turns out that it is error with read group information! Well, re-did the “add read group” step, still NOT working!!
samtools: bam_plcmd.c:596: group_smpl: Assertion `id >= 0 && id n’ failed.
On the recalibrated bam , no_RG case file (without RG due to error)
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b37.sh
2. Input: /nfs/chgv/seqanalysis2/GATK/recalBam/als9c2.build37.recal.noRG.bam
3. OutputDir: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/
4. Submit job: qsub /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b37.sh /nfs/chgv/seqanalysis2/GATK/recalBam/als9c2.build37.recal.noRG.bam /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/
5. Sent May 18th, 2011 @10:25 a.m.; finished by May 23rd @4:49 a.m.
Unfortunately, there has been errors with process, ended up in variant call truncated at chromosome 9! Could be “disk space issue”.
Comparing size of file1:
For no_RG case:
[jl407@head4 noRG_reCal]$ ls -al file1
-rw-r–r– 1 jl407 chavi 264284334703 May 23 04:49 file1
For raw bam case:
[jl407@head4 noRG_reCal]$ ls -al /nfs/chgv/seqanalysis2/ALIGNMENT/samples/als9c2/build37/combined/file1
-rw-r–r– 1 jl407 chavi 264495324652 May 4 08:52 /nfs/chgv/seqanalysis2/ALIGNMENT/samples/als9c2/build37/combined/file1
Compare bam file:
[jl407@head4 noRG_reCal]$ ls -al /nfs/chgv/seqanalysis2/GATK/recalBam/
total 157200748
drwxr-xr-x 2 jl407 chavi 4096 May 18 11:39 .
drwxr-xr-x 11 jl407 chavi 4096 May 23 15:11 ..
-rw-r–r– 1 jl407 chavi 160649148678 May 18 10:13 als9c2.build37.recal.noRG.bam
-rw-r–r– 1 jl407 chavi 8763904 May 18 11:39 als9c2.build37.recal.noRG.bam.bai
[jl407@head4 noRG_reCal]$ ls -al /nfs/chgv/seqanalysis2/ALIGNMENT/samples/als9c2/build37/combined/combined_rmdup.bam
-rw-r–r– 1 jl407 chavi 98071917844 Apr 29 14:23 /nfs/chgv/seqanalysis2/ALIGNMENT/samples/als9c2/build37/combined/combined_rmdup.bam
Need to re-run this, starts on May 24th, @ 4:00 p.m.
Step two: Split vcf files by snps and indels
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl
2. Input: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/var_flt_vcf
3. RunningDir/OutputDir: /nfs/chgv/seqanalysis2/GATK/runningDir
4. cd to the RunningDir, and submit job:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl /nfs/chgv/seqanalysis2/GATK/runningDir/genome_vcf_files, this file contains a list of var_flt_vcf to process.
als9c2 /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/var_flt_vcf
For no_RG case:
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl
2. Input: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/var_flt_vcf
3. RunningDir/OutputDir: /nfs/chgv/seqanalysis2/GATK/runningDir
4. cd to the RunningDir, and submit job:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl /nfs/chgv/seqanalysis2/GATK/runningDir/genome_vcf_files, this file contains a list of var_flt_vcf to process.
als9c2 /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/var_flt_vcf
Step three: Filter vcf variants
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl
2. Input: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/var_flt_vcf.snp
3. RunningDir/OutputDir: /nfs/chgv/seqanalysis2/GATK/runningDir
4. cd to the RunningDir, and submit job:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl /nfs/chgv/seqanalysis2/GATK/runningDir/vcf_snp_filelist snp
vcf_snp_filelist contains a list of var_flt_vcf.snp to process.
als9c2 /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/var_flt_vcf.snp
For no_RG case:
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl
2. Input: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/var_flt_vcf.snp
3. RunningDir/OutputDir: /nfs/chgv/seqanalysis2/GATK/runningDir
4. cd to the RunningDir, and submit job:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl /nfs/chgv/seqanalysis2/GATK/runningDir/vcf_snp_filelist snp
vcf_snp_filelist contains a list of var_flt_vcf.snp to process.
als9c2 /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/var_flt_vcf.snp
Result: should be in the following format, two files with the same name as your variant file will be created:
.filtered (variants that passed QC threshold)
.filtered.out (variants which failed QC)
Step four: ti/tv ratio
Dependency:
Java path: export PATH=/home/csem/tm103/DSCR/jre1.6.0_13/bin:$PATH
GATK package: gatk_dir=/nfs/chgv/seqanalysis/SOFTWARE/GenomeAnalysisTK-1.0.5083
Reference DB (build 37): reference=/nfs/chgv/seqanalysis/ALIGNMENT/BWA_INDEX_JYL/human_g1k_v37.fasta
Commands to issue
1. Samples ( als9c2) dir:/nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/
2. Script :/nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv.sh
3. Log files: /nfs/chgv/seqanalysis2/GATK/logs/
4. Output: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/gatk_titv.csv
5. Command: qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv.sh /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/
For no_RG case: — Changes in Italic
1. Two input information:
a. Samples ( als9c2) dir:/nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/
b. Output file name: /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/als9c2.build37.noRG.recal.gatk_titv.csv
2. Script :/nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv.sh
3. Log files: /nfs/chgv/seqanalysis2/GATK/logs/
4. Command: qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv.sh /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/ als9c2.build37.noRG.recal.gatk_titv.csv
FIXME: this output needs to be “sample” oriented!!!, i.e. als9c2.build37.precal.gatk_titv.csv
Step five: Homozygous/heterozygous SNV ratio
Check to see if the genotype is listed in the 10th column in the VCF file
Script: /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl
Usage: perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl vcf_file > printTO
Procedure: cd to “/nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/” and issue command: perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/var_flt_vcf.snp.filtered > als9c2.build37.precal.homhet.snp.ratio.hets
For noRG_recal case:
Script: /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl
Usage: perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl vcf_file > printTO
Procedure: cd to “/nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/” and issue command: perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/var_flt_vcf.snp.filtered > als9c2.build37.noRG.recal.homhet.snp.ratio.hets
Step six: Concordance check with SVA
(1) After the “variant call” in step one, generate bco files in the DSCR
Use the following 2 lines from this script
/nfs/chgv/seqanalysis2/GATK/scripts/bcftools_coverage_bco.sh:
Modify all the parameters, including Java path, samtools location, reference DB, etc.
export PATH=/home/csem/tm103/DSCR/jre1.6.0_13/bin:$PATH
sam_dir=/nfs/chgv/seqanalysis/SOFTWARE/samtools-0.1.12a
reference_fasta_file=/nfs/chgv/seqanalysis/ALIGNMENT/BWA_INDEX_JYL/human_g1k_v37.fasta
If it runs manually, a few subdirectories need to be created properly.
a. bco_dir=$combined_dir/bco
b. $bco_dir/$sample
c. seqsata_sample_dir=/nfs/testrun/GATK-recal/assessmentComp/samples/$sample (for the results to be transferred back to our sva clusters).
Example command:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/bcftools_coverage_bco_build37.sh /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/ /nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal
/nfs/chgv/seqanalysis/SOFTWARE/samtools-0.1.12a/bcftools/bcftools view -cg /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal//file1 > /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal//bcftools_file
java -jar /nfs/chgv/seqanalysis2/GATK/scripts/vcf2bco.jar /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal//bcftools_file /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal//bco/
rsync -a –partial –timeout=10000 -e ‘ssh’ -r /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal//bco sva6.lsrc.duke.edu:/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal
For noRG_case:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/bcftools_coverage_bco_build37.sh /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/ /nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/noRG_reCal
/nfs/chgv/seqanalysis/SOFTWARE/samtools-0.1.12a/bcftools/bcftools view -cg /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal//file1 > /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal//bcftools_file
java -jar /nfs/chgv/seqanalysis2/GATK/scripts/vcf2bco.jar /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal//bcftools_file /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal//bco/
rsync -a –partial –timeout=10000 -e ‘ssh’ -r /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal//bco sva6.lsrc.duke.edu:/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/noRG_reCal
At last step, those bco files should be transferred back to LSRC
(2) Create an SVA project (meaning get a .gsap file created)
Since this is alignment against build 37, there are many changes to the sva project. Thanks to Dongliang, who helped me with the set ups:
Currently, The new build is existing at: /nfs/goldstein/goldsteinlab/software/SequenceVariantAnalyzer_1.1_b37/
The .gsap file needs to be modified accordingly:
1. Menu File -> Open example project file
2. Copy all the lines after line “#Reference database version”
3. Replace the lines in the same place in your build 36 .gsap project file 4. Remove all the starting “#” in the “[REFBIN]=” lines. Because your project has all chromosomes, while the example only has X.
4. At last, Dongliang had to modified the permission for ./datasource/ensembl/61_37f/variation_feature2.txt
MODIFIED .gsp file!! May 18th, 2011
.GSAP file for SVA project: /nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal/vcf_als9c2_precal.gsap
(3) Topstrand file names for als9c2:
/nfs/testrun/GATK-recal/svaprojects/vcf/genome/topstrandfilenames (Illumina created/provided this file)
/nfs/testrun/GATK-recal/svaprojects/vcf/genome/topstrandfilenames, the content in this file is as following.
SAMPLE_ID GENOTYPING_ID TopStrand_files
als9c2, als009c2, /nfs/sva01/SequenceAnalyses/TopStrandFiles/als006-015_10samples__MAY_4_10_TopStrand.csv
(4) Use this version of SVA: (It is about 120 mitues.
/nfs/goldstein/goldsteinlab/software/SequenceVariantAnalyzer_1.1_b37/
(Make sure to uncheck the filtering option in sva)
With Xwin, I was able to ssh to sva0.lsrc.duke.edu and perform the “annotation” . Started @7:30 a.m. May 19th, 2011.
(5) Check concordance:
SVA version: /nfs/goldstein/goldsteinlab/software/SequenceVariantAnalyzer_1.1_b37/
Case I: Once “annotate” is done, go directly to the following steps to get concordance.
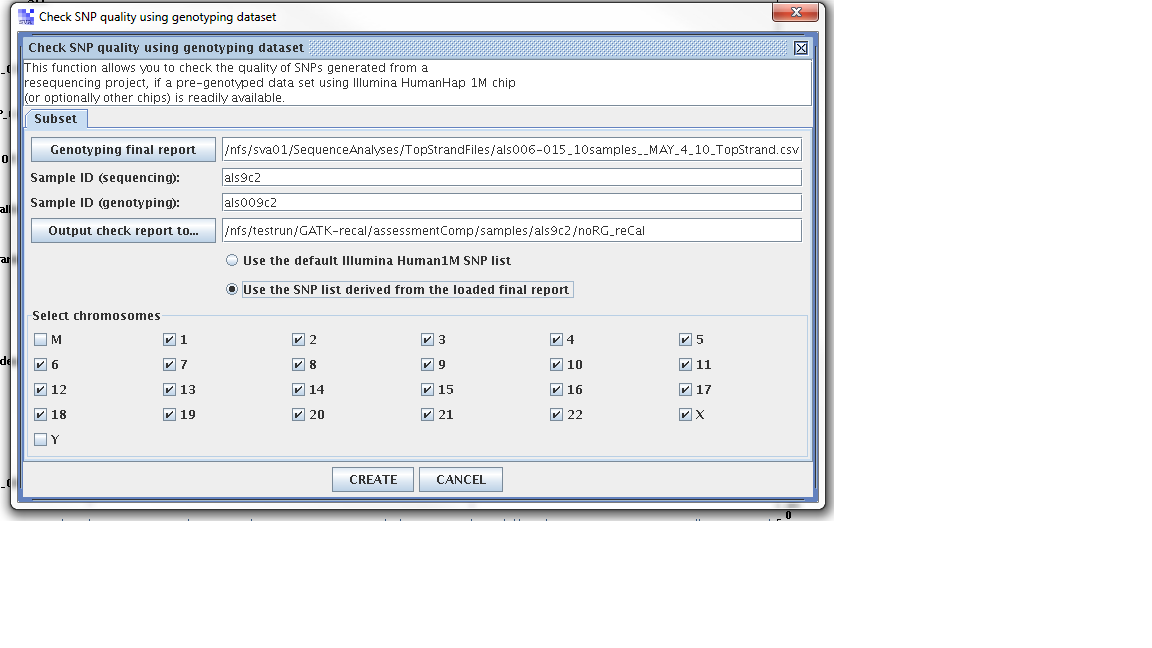
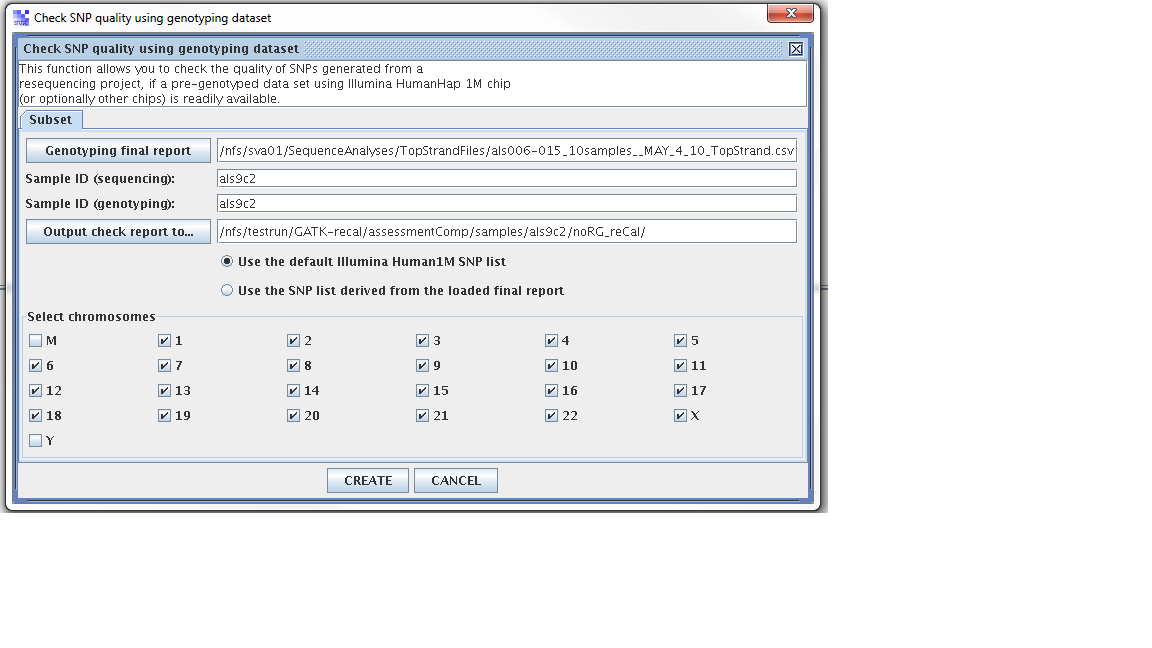
Step 1. From the SVA UI, choose “summary -> check SNP quality using genotyping dataset”
Step 2: Filling in information for four fields, information is stored at
Four fields
Genoptying final report: //nfs/sva01/SequenceAnalyses/TopStrandFiles/als006-015_10samples__MAY_4_10_TopStrand.csv
sample ID (sequencing): als9c2
sample ID (genotyping): als009c2
output check report to: /nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal
Step 3: Check “use the SNP list derived from the loaded final report”
Step 4: Click “Create”
Case II: If the project is closed, load the project by clicking “load” button and go back to the steps mentioned above.
Results:
For pre-calibration case
Performing quality check using genotype data (user SNP list).
> Sample ID (sequencing): als9c2
Sample ID (genotyping): als009c2
Note: 584882 SNPs are loaded from user SNP report.
– Chromosome: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X
– 297962 SNPs (with rs# listed in genotype data (user SNP list) are loaded from your resequencing project.
– 148881 SNPs on bottom strand (Illumina).
– Among these, 296597 SNPs are found in genotype data (user SNP list) .
– Among the 297962 SNPs loaded from resequencing project, there are:
294755 match with genotype data (user SNP list);
1842 mismatch;
1365 not found in genotype data (user SNP list).
– Among the 1842 mismatches, there are
21 hom (seq) – hom (geno)
32 hom (seq) – het (geno)
18 het (seq) – het (geno)
1771 het (seq) – hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal.nomatch.txt
[Part 2] N = 286770
– Among the 286770 SNPs included in the genotyping chip but not derived from sequencing, there are:
286767 that can be correctly mapped to reference sequence;
Among these 286767 there are 281402 matches;
and 5365 mismatches, including 3699 het (geno) and 1666 hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal.part2.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/preCal.part2.nomatch.txt
Results for recalibrated results

Performing quality check using genotype data (user SNP list).
> Sample ID (sequencing): als9c2
Sample ID (genotyping): als009c2
Note: 584882 SNPs are loaded from user SNP report.
– Chromosome: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X
– 299193 SNPs (with rs# listed in genotype data (user SNP list) are loaded from your resequencing project.
– 149493 SNPs on bottom strand (Illumina).
– Among these, 297794 SNPs are found in genotype data (user SNP list) .
– Among the 299193 SNPs loaded from resequencing project, there are:
296497 match with genotype data (user SNP list);
1297 mismatch;
1399 not found in genotype data (user SNP list).
– Among the 1297 mismatches, there are
21 hom (seq) – hom (geno)
32 hom (seq) – het (geno)
20 het (seq) – het (geno)
1224 het (seq) – hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/noRG_reCal.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/noRG_reCal.nomatch.txt
[Part 2] N = 285979
– Among the 285979 SNPs included in the genotyping chip but not derived from sequencing, there are:
282538 that can be correctly mapped to reference sequence;
Among these 282538 there are 138818 matches;
and 143720 mismatches, including 2429 het (geno) and 141291 hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/noRG_reCal.part2.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/als9c2/noRG_reCal.part2.nomatch.txt
As the results show, we see improvement on the concordance with both part I and part II. And, it is more important that we see improvement with part II.
So the question exists, why/what causes low concordance check with genotyp chip results???
Step seven: Overlap of filtered autosomal SNVs with dbsnp
Here we need a little bit more attention. Since we are trying to compare analysis against build 37, we need snp information for build 37. It is available at NCBI/dbsnp
Download dbsnp file as following:
1. Download b132_SNPChrPosOnRef_37_1.bcp.gz
2. Check download completeness
[jl407@head4 refDB]$ md5sum b132_SNPChrPosOnRef_37_1.bcp.gz
07acf58908cf857b62e9de99a1d56533 b132_SNPChrPosOnRef_37_1.bcp.gz
[jl407@head4 refDB]$ cat b132_SNPChrPosOnRef_37_1.bcp.gz.md5
MD5(b132_SNPChrPosOnRef_37_1.bcp.gz)= 07acf58908cf857b62e9de99a1d56533
1. Samples (info) located: /nfs/chgv/seqanalysis2/GATK/doc/snplistFile
2. Script (dbsnp_overlap.pl) located: /nfs/chgv/seqanalysis2/GATK/scripts/
3. Log files: /nfs/chgv/seqanalysis2/GATK/logs/
4. Command: qsub /nfs/chgv/seqanalysis2/GATK/scripts/dbsnp_overlap_b37.pl /nfs/chgv/seqanalysis2/GATK/doc/snplistFile
5. Results were written to log file
For no_RG case:
The only change is where the var_flt_vcf.snp.filtered is located:
als9c2 /nfs/chgv/seqanalysis2/GATK/samples/als9c2/preCal/var_flt_vcf.snp.filtered
als9c2 /nfs/chgv/seqanalysis2/GATK/samples/als9c2/noRG_reCal/var_flt_vcf.snp.filtered
Here is the concordance checking results: concordance_als9c2_b37
July 19th, 2011
Wanted to test “using 1M vs snp loaded from the project”.