
This is the assessment on GATK recalibration, using our current variant calling pipeline.
Samples aligned to build36
dukscz0106
mchd002A2
na12878
Major steps:
Step 1: calling variants with samtools1.0.12a
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b36.sh
2. Input: /nfs/chgv/seqanalysis2/GATK/recalBam/dukscz0106.build36.recal.noRG.bam
3. OutputDir: /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/
4. Submit job: qsub /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b36.sh /nfs/chgv/seqanalysis2/GATK/recalBam/dukscz0106.build36.recal.noRG.bam /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/
5. Sent @7:50 a.m. June 11th, but the queue was very long!For mchd002A2: qsub /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b36.sh /nfs/chgv/seqanalysis2/GATK/recalBam/mchd002A2.build36.recal.noRG.bam /nfs/chgv/seqanalysis2/GATK/samples/mchd002A2/noRG_recal/
For na12878, run on lsrc and results were transferred back to dscr.
For comparison purposed, run one on the dscr as well @1:55 p.m. June 23rd, 2010: qsub /nfs/chgv/seqanalysis2/GATK/scripts/call_var_from_bam_b36.sh /nfs/chgv/seqanalysis2/GATK/recalBam/na12878.build36.recal.noRG.bam /nfs/chgv/seqanalysis2/GATK/samples/na12878/noRG_reCal/
Here are a few QC metrics we can check
1. ti/tv ratio
2. number of raw and filtered SNV
3. number of indel called by samtools
4. proportion of heterozygous SNVs on the x chr compared to the total number of SNVs in the X
5. Ratio of homo to hetero SNVs
Split vcf files by snps and indels
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl
2. Input: /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/var_flt_vcf (var_raw_bcf is binary file, where var_flt_vcf was generated from), also, this “var_flt_vcf” is really “RAW” SNV/INDEL file.
3. RunningDir/OutputDir: /nfs/chgv/seqanalysis2/GATK/runningDir
4. cd to the RunningDir, and submit job:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl /nfs/chgv/seqanalysis2/GATK/runningDir/dukscz0106_vcf_files , this file contains a var_flt_vcf file to process.dukscz0106 /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/var_flt_vcf
5. For mchd002A2: qsub /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl /nfs/chgv/seqanalysis2/GATK/runningDir/mchd002A2_vcf_files
6. For na12878: qsub /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl /nfs/chgv/seqanalysis2/GATK/runningDir/na12878_vcf_files
7. For na12878_raw: qsub /nfs/chgv/seqanalysis2/GATK/scripts/split_snps_indels.pl /nfs/chgv/seqanalysis2/GATK/samples/na12878/no_reCal/na12878_noGATK_vcf_files
Filter vcf variants
/nfs/chgv/seqanalysis/ALIGNMENT/QC/Scripts/sam12/vcf_varfilter.pl
Your variant list should be in the format;
Two files with the same name as your variant file will be created:
.filtered (variants that passed QC threshold)
.filtered.out (variants which failed QC)
Example usage:
qsub /nfs/chgv/seqanalysis/ALIGNMENT/QC/Scripts/sam12/vcf_varfilter.pl /nfs/chgv/seqanalysis/ALIGNMENT/QC/samtools_version/sam12/genome/vcf_snp_filelist snp
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl
2. Input: /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/var_flt_vcf.snp
3. RunningDir/OutputDir: /nfs/chgv/seqanalysis2/GATK/runningDir
4. cd to the RunningDir, and submit job:
qsub /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl /nfs/chgv/seqanalysis2/GATK/runningDir/vcf_snp_dukscz0106 snp/,
vcf_snp_filelist contains a list of var_flt_vcf.snp to process.dukscz0106 /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/var_flt_vcf.snp
5. For mchd002A2: qsub /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl /nfs/chgv/seqanalysis2/GATK/runningDir/vcf_snp_mchd002A2 snp
6. For na12878: qsub /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl /nfs/chgv/seqanalysis2/GATK/runningDir/vcf_snp_na12878 snp
7. For na12878_raw: qsub /nfs/chgv/seqanalysis2/GATK/scripts/vcf_varfilter.pl /nfs/chgv/seqanalysis2/GATK/samples/na12878/no_reCal//vcf_snp_na12878_raw snp
ti/tv ratio
1. Samples ( dukscz0106 & mchd002A2) located: /nfs/chgv/seqanalysis2/GATK/samples/
2. Script (gatk_titv_b36.sh) located: /nfs/chgv/seqanalysis2/GATK/scripts/
3. Log files: /nfs/chgv/seqanalysis2/GATK/logs/
4. Usage: qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv_b36.sh /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal dukscz0106.gatk_titv.csv
5. For mchd002A2: qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv_b36.sh /nfs/chgv/seqanalysis2/GATK/samples/mchd002A2/noRG_recal/ mchd002A2.gatk_titv.csv
6. For na12878: qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv_b36.sh /nfs/chgv/seqanalysis2/GATK/samples/na12878/noRG_reCal_lsrc/ na12878.gatk_titv.csv7. For na12878_raw: qsub /nfs/chgv/seqanalysis2/GATK/scripts/gatk_titv_b36.sh /nfs/chgv/seqanalysis2/GATK/samples/na12878/no_reCal/ na12878.gatk_titv.csv
Homozygous/heterozygous SNV ratio
Check to see if the genotype is listed in the 10th column in the VCF file
Script: /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl
Usage: perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl vcf_file > printTO
Example: perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl var_flt_vcf.snp.filtered > vcf.snp.filtered.hets
Usage: cd to /workingDir/ and run the script:
perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl var_flt_vcf.snp.filtered > dukscz0106.hets
perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl var_flt_vcf.snp.filtered > mchd002A2.hets
perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl var_flt_vcf.snp.filtered > na12878.hets
perl /nfs/chgv/seqanalysis2/GATK/scripts/vcf_hets.pl var_flt_vcf.snp.filtered > na12878_raw.hets
Step 2: Generate SVA project with samtools1.0.12a
1. Script: /nfs/chgv/seqanalysis2/GATK/scripts/bcftools_coverage_bco_build36.sh
2. Input(dscr): /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/file1
3. OutDir(lsrc)r: /nfs/testrun/GATK-recal/assessmentComp/samples/dukscz0106/noRG_reCal/
4. Submit job: qsub /nfs/chgv/seqanalysis2/GATK/scripts/bcftools_coverage_bco_build36.sh dukscz0106 /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/ /nfs/testrun/GATK-recal/assessmentComp/samples/dukscz0106/noRG_reCal/5. Job for mchd002A2: qsub /nfs/chgv/seqanalysis2/GATK/scripts/bcftools_coverage_bco_build36.sh mchd002A2 /nfs/chgv/seqanalysis2/GATK/samples/mchd002A2/noRG_recal/ /nfs/testrun/GATK-recal/assessmentComp/samples/mchd002A2/noRG_reCal
For dukscz0106, sent @12:05 p.m. June 11th!
For mchd002a2, sent @8:36 p.m. June 20th!
Here we can check:
6. concordance of SNVs with genotyping chip computed with SVA
Step 3. Overlap of filtered autosomal SNVs with dbsnp
1. Samples ( dukscz0106 ) located: /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/noRG_reCal/var_flt_vcf.snp.filtered
2. Script (dbsnp_overlap.pl) located: /nfs/chgv/seqanalysis2/GATK/scripts/dbsnp_overlap.pl
3. Log files: /nfs/chgv/seqanalysis2/GATK/logs/
4. Command: qsub /nfs/chgv/seqanalysis2/GATK/scripts/dbsnp_overlap.pl /nfs/chgv/seqanalysis2/GATK/doc/dukscz0106.snp
5. For mchd002A2: qsub /nfs/chgv/seqanalysis2/GATK/scripts/dbsnp_overlap.pl /nfs/chgv/seqanalysis2/GATK/doc/mchd002A2.snp
6. For na12878: qsub /nfs/chgv/seqanalysis2/GATK/scripts/dbsnp_overlap.pl /nfs/chgv/seqanalysis2/GATK/doc/na12878.snp
7. Results were written to log file
7. overlap of filtered autosomal SNVs with dbsnp
Learning from Jessica on variant call
Concordance check with SVA
(1) Generate bco files in the DSCR
Use the following 2 lines from this script
/nfs/chgv/seqanalysis2/GATK/scripts/bcftools_coverage_bco.sh:
Modify all the parameters, including Java path, samtools location, reference DB, etc.
If it runs manually, a few subdirectories need to be created properly.
a. bco_dir=$combined_dir/bco
b. $bco_dir/$sample
c. seqsata_sample_dir=/nfs/testrun/GATK-recal/assessmentComp/samples/$sample (for the results to be transferred back to our sva clusters).
Note: For mchd002A2, job sent May 2nd, @ 11:00 a.m. and finishes @ 1:02 a.m. May 3rd
Note: For dukscz0106, job sent May 3rd, @ 11:16 a.m. and finishes @ 3:30 a.m. May 4th
Usage: qsub bcftools_coverage_bco_jyl.sh /nfs/chgv/seqanalysis2/GATK/samples/dukscz0106/postCal dukscz0106
At last step, those bco files should be transferred back to LSRC
(2) Create an SVA project (meaning get a .gsap file created)
Example of SVA project:
/nfs/svaprojects/vcf/genome/vcf_genomes_3.gsap
Details see the explanation on the .gsap file
/nfs/testrun/GATK-recal/assessmentComp/samples/mchd002A2/vcf_mchd002A2_recal.gsap
/nfs/testrun/GATK-recal/assessmentComp/samples/dukscz0106/vcf_dukscz0106_recal.gsap
(3) Topstrand file names for the 2 samples:
/nfs/svaprojects/vcf/genome/topstrandfilenames (Illumina created/provided this file)
/nfs/testrun/GATK-recal/svaprojects/vcf/genome/topstrandfilenames, the content in this file is as following. The original copy was saved and individual one is used per sample.
mchd002A2, MCHD002, /nfs/sva01/SequenceAnalyses/TopStrandFiles/MCHD_Family__FEB_11_2010_TOP_FinalReport.csv
dukscz0106, Duk-Schiz0106DEF, /nfs/sva01/SequenceAnalyses/TopStrandFiles/Duke_Scz_16_samples__DEC_09_09_FinalReport_TOP.csv
(4) Use this version of SVA: (It is about 120 mitues.
/nfs/goldstein/goldsteinlab/software/SequenceVariantAnalyzer_1.1 (how to use this program??)
(Make sure to uncheck the filtering option in sva)
Step 1. Grab any machine along the hallway and log in with netID/password
Step 2. Make sure that the drive has been mounted
Step 3. cd to the directory /nfs/goldstein/goldsteinlab/software/SequenceVariantAnalyzer_1.1/, as this is the version for new VCF format
Step 4. Lunch SVA, ./SequenceVariantAnalysis.sh
Step 5. From File -> Open, explore through to the directory that contains that .gsap file, make sure that all the parameters are set up properly
Step 6. Clicking “Annotating”
(5) Check concordance:
SVA version: /nfs/goldstein/goldsteinlab/software/SequenceVariantAnalyzer_1.1/
Case I: Once “annotate” is done, go directly to the following steps to get concordance.
Step 1. From the SVA UI, choose “summary -> check SNP quality using genotyping dataset”
Step 2: Filling in information for four fields, information is stored at
/nfs/testrun/GATK-recal/svaprojects/vcf/genome/opstrandfilenames, the content in this file is:
mchd002A2, MCHD002, /nfs/sva01/SequenceAnalyses/TopStrandFiles/MCHD_Family__FEB_11_2010_TOP_FinalReport.csv
Four fields
Genoptying final report: /nfs/sva01/SequenceAnalyses/TopStrandFiles/MCHD_Family__FEB_11_2010_TOP_FinalReport.csv
sample ID (sequencing): mchd002A2
sample ID (genotyping): MCHD002
output check report to: /nfs/testrun/GATK-recal/assessmentComp/samples/mchd002A2
Step 3: Check “use the SNP list derived from the loaded final report”
Step 4: Click “Create”
Case II: If the project is closed, load the project by clicking “load” button and go back to the steps mentioned above.
Results:
Performing quality check using genotype data (user SNP list).
> Sample ID (sequencing): mchd002A2
Sample ID (genotyping): MCHD002
Note: 584882 SNPs are loaded from user SNP report.
– Chromosome: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X
– 300585 SNPs (with rs# listed in genotype data (user SNP list) are loaded from your resequencing project.
– 150265 SNPs on bottom strand (Illumina).
– Among these, 300351 SNPs are found in genotype data (user SNP list) .
– Among the 300585 SNPs loaded from resequencing project, there are:
299325 match with genotype data (user SNP list);
1026 mismatch;
234 not found in genotype data (user SNP list).
– Among the 1026 mismatches, there are
16 hom (seq) – hom (geno)
34 hom (seq) – het (geno)
21 het (seq) – het (geno)
955 het (seq) – hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/mchd002A2/noRG_reCal.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/mchd002A2/noRG_reCal.nomatch.txt
[Part 2] N = 283814
– Among the 283814 SNPs included in the genotyping chip but not derived from sequencing, there are:
283814 that can be correctly mapped to reference sequence;
Among these 283814 there are 280139 matches;
and 3675 mismatches, including 2798 het (geno) and 877 hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/mchd002A2/noRG_reCal.part2.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/mchd002A2/noRG_reCal.part2.nomatch.txt
For sample dukscz0106, the cooncordance check results are
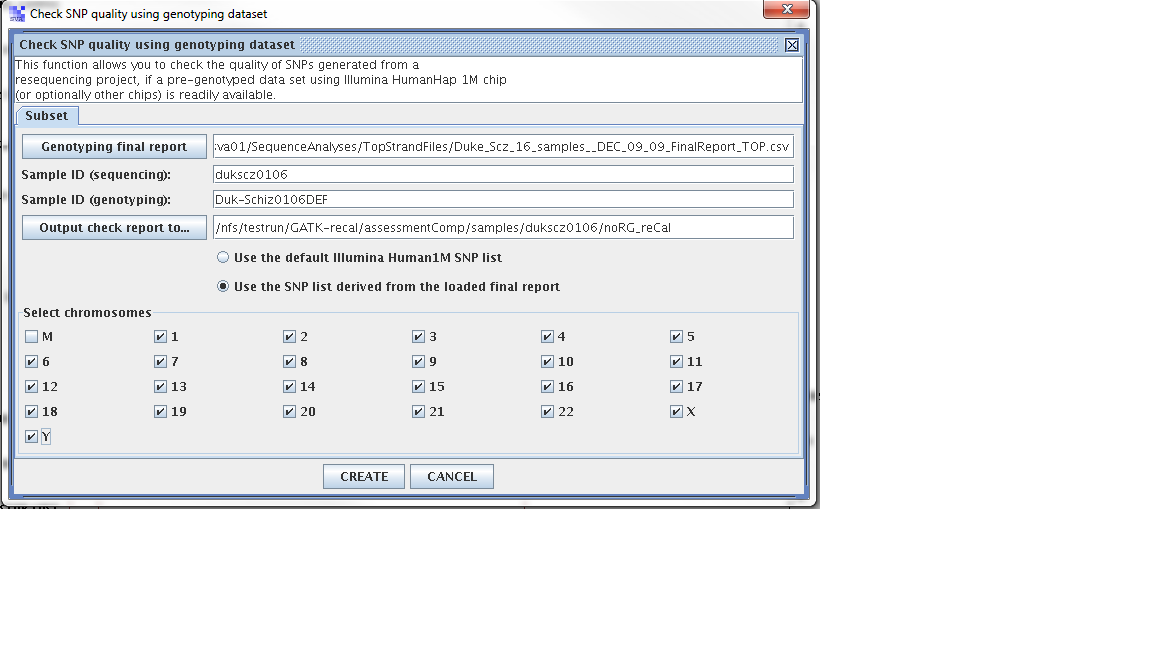
Performing quality check using genotype data (user SNP list).
> Sample ID (sequencing): dukscz0106
Sample ID (genotyping): Duk-Schiz0106DEF
Note: 583754 SNPs are loaded from user SNP report.
– Chromosome: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X
– 297679 SNPs (with rs# listed in genotype data (user SNP list) are loaded from your resequencing project.
– 148586 SNPs on bottom strand (Illumina).
– Among these, 297646 SNPs are found in genotype data (user SNP list) .
– Among the 297679 SNPs loaded from resequencing project, there are:
294629 match with genotype data (user SNP list);
3017 mismatch;
33 not found in genotype data (user SNP list).
– Among the 3017 mismatches, there are
20 hom (seq) – hom (geno)
124 hom (seq) – het (geno)
88 het (seq) – het (geno)
2785 het (seq) – hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/dukscz0106/noRG_reCal.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/dukscz0106/noRG_reCal.nomatch.txt
[Part 2] N = 287131
– Among the 287131 SNPs included in the genotyping chip but not derived from sequencing, there are:
287128 that can be correctly mapped to reference sequence;
Among these 287128 there are 280499 matches;
and 6629 mismatches, including 5652 het (geno) and 977 hom (geno)
Detailed lists were written to:
/nfs/testrun/GATK-recal/assessmentComp/samples/dukscz0106/noRG_reCal.part2.match.txt
/nfs/testrun/GATK-recal/assessmentComp/samples/dukscz0106/noRG_reCal.part2.nomatch.txt
concordance ratio = (294629+280499)/(297646+287128) = 0.9835
QC metrics checked: comparison_to_report_JM
SVA concordance only: concordance_comparison
To compare details concordance checked done on raw bam file, all the sva project logs are located: /nfs/svaprojects/vcf/genome/concordance
For na12878 sample, we don’t have the genotype array data to compare to (as we did with in house samples). However, we do have the 1000 genome snp releases.
Step 1. Download the 1000 genome release
Step 2. Compare our in-house vcf file to the 1000 genome release.
Script located: /nfs/chgv/seqanalysis2/GATK/scripts
Script usage: qsub /nfs/chgv/seqanalysis2/GATK/scripts /dir/ /dir/
Script output: Results will be printed to log file.
Step 3. Comparison results are here: concordance_na12878