The Itchy Sheep Hypothesis
To begin, it may seem puzzling that scientists would even suspect the existence of misshapen proteins as the cause of infectious disease. What led to this theory in the first place? The answer, it turns out, is sheep. For centuries, shepherds have sometimes noticed peculiar behavior in their flocks – a formerly healthy sheep will begin to lose weight and have difficulty walking about its pen. The sick animal also begins to rub against fence posts or other rough surfaces as if to alleviate an unbearable itch. The disease – scrapie – actually gets its name from this scraping movement4,5. Eventually, the animal becomes paralyzed and succumbs to starvation6.

Traditionally, scientists thought the disease only occurs in sheep with a inherent genetic weakness that leads them to develop scrapie when exposed to the prions we will discuss later. However, recent research suggests that, in addition to this genetic predisposition, scrapie can be caused by truly infectious prions that effect even flocks thought to be genetically resistant to scrapie8. Generally, sheep acquire the disease by ingesting prion particles in their environment9. Also, these particles can be transmitted between members of a flock, or prenatally from mothers to their young10.

After the sheep’s death, brain dissection would show that the animal’s brain and other neural tissue were decayed5. Because the sheep’s nervous system malfunctions as the individual neurons die, sensory signals are unable to successfully make their way from the skin to the brain. As a result of its deteriorating nervous system, the scrapie-infected sheep does not respond normally to stimuli such as pressure and begins to uncontrollably itch5.
But what had caused the sheep’s brain to deteriorate in the first place? When scientists first began to study scrapie, they had immense trouble tracking down an origin. Hoping to isolate a pathogen from tissue in scrapie-infected sheep, they employed a common test called filtration. As we have learned throughout this course, viruses are much smaller in size than bacteria, parasites, and other infectious pathogens. Consequently, a tissue sample (like blood) from a bacterial infection would be noninfectious if it was first filtered through a membrane with pores small enough to allow only viruses to pass. In other words, the filtrate (or solution collected after filtration) would no longer contain the bacteria or parasites because the pores would have obstructed their passage. Using this technique, scientists found that the filtrates remained infectious even after being passed through the membrane, suggesting that bacteria or larger parasites were not to blame. Perhaps viruses were the cause?
However, viruses need genetic material such as DNA or RNA to replicate. Using this knowledge, scientists next treated scrapie-infected tissues with chemicals that destroy nucleic acids and then examined the tissue’s infective nature3. Surprisingly, the tissue remained infectious, suggesting that a viral cause was unlikely as well. If the cause was not bacterial nor parasitic nor viral, what could it be?
Despite a lack of an identifiable pathogen, scrapie can still be transmitted by blood transfusion between sheep, suggesting that something is there. One clue to a cause, however, may be the misshapen protein clusters that are always present in the neural tissue of scrapie-infected sheep. In fact, these clusters must be present in a tissue sample in order to be infectious5. So, scientists next applied chemicals that destroy proteins to the scrapie-infected tissue. The result: the tissue could no longer transmit scrapie. With this treatment, the infectious nature of the unidentified “pathogen” had been lost5. But could this mean that a protein could be to blame?
The Prion Particle
Originally, the idea that a protein could cause disease was thought impossible. The infectious agent responsible for scrapie was simply known as the “scrapie agent.” Scientists did not know what it was, but assumed that a bacteria or virus was involved11. The most that researchers were willing to admit was that experiments showed a protein was associated with this “scrapie agent,” without suggesting that the protein could cause disease by itself11. It took a bold leap by future Nobel Laureate Stanley Prusiner to suggest that, indeed, a protein could cause disease, and he created the term “prion” to describe this unprecedented pathogen3.
The Mysterious PrP
Normally, the prion protein (or PrP) adheres to the plasma membrane of neurons by a sugar anchor12,13. In this position, it is thought to help transmit chemical signals between adjacent nervous cells as a part of the normal process by which a sensory experience (be it touch, taste, smell, or otherwise) is turned into the electrochemical language read by the brain. When unneeded, PrP is degraded by proteases, the protein-cleaving enzymes discussed in previous units15.

However, normal PrP can undergo a structural change to generate a disease-causing form (called PrPsc for its ability to now cause scrapie). Though the amino acid sequences of PrP and PrPsc are identical, the two proteins differ significantly in their three-dimensional structure. Experimental evidence has confirmed this: PrP consists mostly of alpha-helices while PrPsc is composed mainly of beta-sheets15. Once formed, a PrPsc can propagate the formation of additional PrPscs16. Just like a single unstable domino can knock over an entire domino line, one PrPsc can convert normal PrPs into aberrant forms16. At each step, the new PrPsc can convert more PrP just as each fallen domino can knock over other dominoes.
Importantly, because of this structural change, PrPsc can no longer be harmlessly cleaved by proteases and consequently begins to accumulate. Because of their abnormal shape, PrPsc proteins tend to stick to one another, and over time, the PrPsc molecules cluster to form long chains called amyloid fibers7. These protein clusters are toxic to neurons, causing neuronal death and ultimately the neurodegeneration seen in scrapie-infected sheep7. A nice animation of this process can be viewed at:

http://mt.gov/liv/animalhealth/diseases/BSE/pathology.asp.
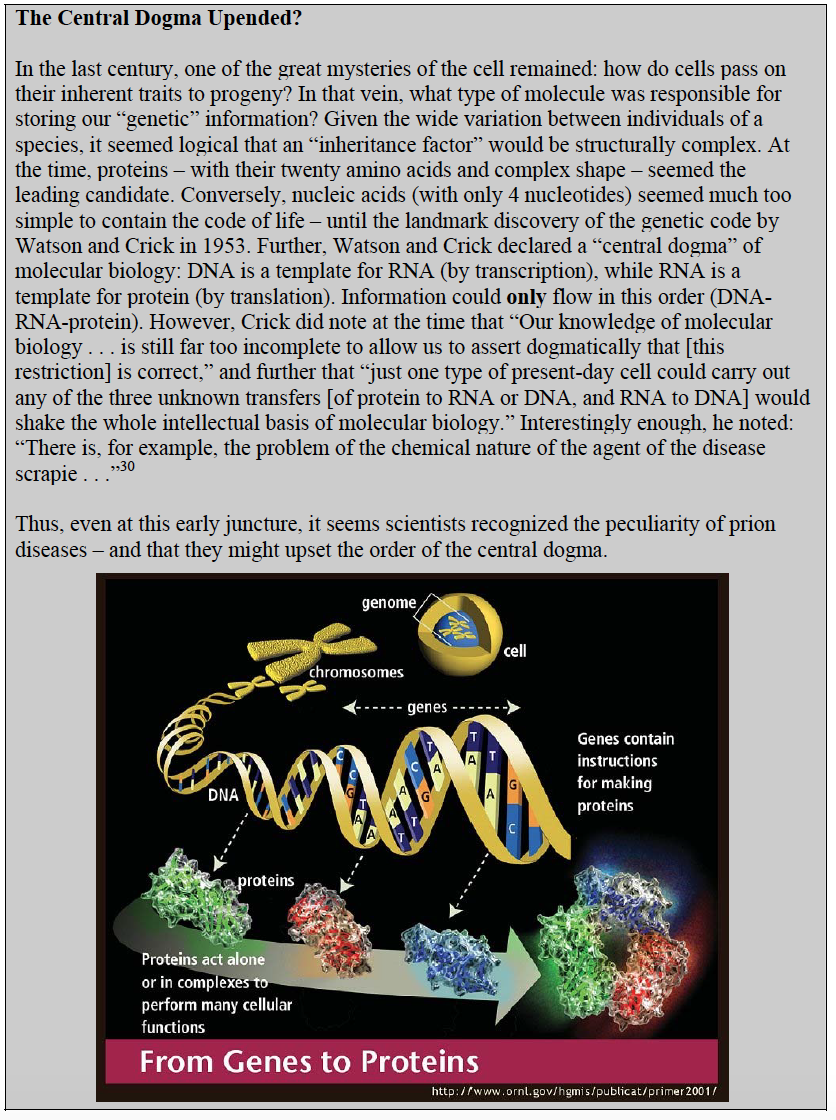
The idea that a change in protein structure causes disease is not new. As we saw in the malaria unit, sickle cell anemia is the result of misfolded hemoglobin. What is unexpected about PrP, however, is that different forms of the disease may be caused by different “misfoldings” – in other words, the 3-D structure of PrP “encodes” its disease-causing properties15. In a world where information of this kind is traditionally maintained in DNA or RNA, this is a radical notion.
Beyond Nucleic Acid: A Protein-Only Disease?
Going back to our investigation, how did scientists determine that proteins could be infectious? Remember the evidence thus far15:
1. The infectious agent is smaller than a bacterium.
2. The infectious agent is not destroyed by chemicals that disrupt nucleic acids.
3. Chemicals that destroy proteins are able to render infectious scrapie tissue noninfectious.
The idea of a protein disease was actually first proposed in 1967, predating the discovery of prions by many years19. At this time it was also declared absurd, but further evidence suggests that proteins may in fact be able to copy themselves in a limited sense. One theory for the formation of PrPsc, for example, is that a PrPsc molecule acts as a template for a catalyzed conversion of PrP to PrPsc – a similar idea in some ways to the replication of DNA16,20,21,22.
Given that PrPsc is always found in scrapie-infected sheep, the disease could be caused by an infectious protein. While this is currently the leading theory for the cause of scrapie and other prion diseases, many scientists have a hard time accepting that a protein can be infectious, and believe that a small and peculiar virus is responsible for these diseases. Proponents of this latter theory claim that the nucleic acid is simply tightly bound to PrP – thus immune to nucleic-acid destroying processes – but lost during the purification process20. Another argument against the “protein-only” theory is that there exist different “strains” of scrapie in laboratory mice, distinguished by incubation time and the tissues where most symptoms occur20. These types of differences are usually encoded in viruses and bacteria through genetic differences. On the other hand, prion theory supporters claim that differences in PrP folding account for these different “strains”16.
The main argument against the “protein-only” hypothesis is that it is so unprecedented. No life form, including small infectious agents such as bacteria and viruses, has ever been discovered that does not use nucleic acids to store information. Thus, it seems impossible that an infectious agent could exist without DNA or RNA. However, there exists much evidence to support the “protein-only” hypothesis15:
1. By changing the amino acid sequence or three-dimensional conformation of PrP, scientists can generate different version of prion diseases in laboratory animals.
2. PrPsc converts PrP to PrPsc in a test tube.
3. Mice lacking the PrP gene cannot become infected with scrapie, but transplanting brain cells with normal PrP into these PrP-deficient mice allows them to develop the disease. Similarly, experiments with mice and hamsters have shown that mice genetically modified to produce the hamster form of PrP can develop scrapie if infected with hamster PrPsc. Otherwise, hamster PrPsc has no effect on the mice.
The experiments in (3) raise the question of whether prion diseases could be genetic as well as infectious. If possessing a certain gene allows a mouse to acquire a prion disease (and lacking that gene makes it immune), it appears that the disease might have some genetic basis. However, it has been shown that sheep that are genetically capable of acquiring scrapie do not do so in a sterile environment15. Therefore, it would appear that external factors cause the disease while genetics can only predispose a sheep to develop scrapie.
Prion Diseases
We have just examined what we know about prion diseases through studies on scrapie. However, prion diseases occur in many other animals including humans.
What is TSE?
One important term to define is TSE, an acronym for Transmissable Spongiform Encephalopathy and the medical term for prion diseases. Encephalopathy means “disease of the brain,” and spongiform refers to the fact that, in prion diseases, the brain deteriorates in a pattern that resembles swiss cheese or a sponge. Thus, TSE is simply a fancy term describing a brain deteriorating illness that can be passed between animals or humans.

Animal Forms of TSE
While the classic example of a prion disease is scrapie, such diseases exist in other species as well23. Perhaps the most famous instance in the current media is Bovine Spongiform Encephalopathy (BSE), the disease more commonly known as “Mad Cow Disease”23. Another notable animal TSE is Chronic Wasting Disease (CWD), which afflicts hoofed mammals in North America including deer24. However, unlike BSE, it is not known to be transmissible to humans. Prion infections have also been observed in mink, goats, and other captive animals24.
Human Forms of TSE
In general, human prion diseases cause progressive loss of motor control, dementia (severe loss of memory or mental function), paralysis, and wasting (a great loss of body and muscle mass, in this case). TSE can also be accompanied by secondary infections including pneumonia.
There are many human diseases caused by prions. The most common is Creutzfeldt-Jacob Disease (CJD), a neurodegenerative disorder that occurs in both spontaneous (non-genetic) and genetically linked forms25. Even in the more common spontaneous form, CJD is relatively rare, afflicting only about 1 person in a million each year worldwide25. As with other neurodegenerative conditions like Alzheimer’s disease, older patients are more likely to develop CJD25. For example, beyond age 50, the incidence of spontaneous CJD is approximately 3.4 per million individuals a year25. The spontaneous disease accounts for 85% of all reports of CJD25.
The rarer genetically-linked form of CJD accounts for only about 15% of cases. It is classified as autosomal dominant condition (i.e. it is due to a mutation in body cells, not germline cells, and only one copy is required to have a negative effect)25. Like familial Alzheimer’s disease, genetically linked CJD affects younger individuals than the spontaneous disease. Genetically-linked CJD is further subdivided based on clinical symptoms,

including forms such as Gerstmann-Straussler-Scheinker (GSS) and fatal familial insomnia. GSS is extremely rare, with an incidence of between 1 and 10 per 100 million per year, and is believed to be caused by a point mutation in the amino acid sequence of PrP26,27. Notably, there are some populations demonstrating higher than average incidence of CJD, like Libyan-born Israeli Jews, for whom the incidence of the disease is 30 per million per year5. A genetic defect can also cause the rare familial fatal insomnia, a prion disease that usually afflicts related individuals. What begins as an inability to sleep progresses to loss of mobility reminiscent of Parkinson’s disease, followed by a loss of mental function and, within 1-3 years, death. Like other prion diseases, there is no known cure28.
There is also a variant form of CJD, denoted vCJD, which was first described in 1996 in the United Kingdom in connection with mad cow disease5. There is now strong scientific evidence that the same agent that causes mad cow also is responsible for vCJD in humans, especially since the only common trait that the vCJD victims in Britain appear to share is that they ate beef5.
Another notable human prion disease is Kuru, a neurodegenerative disorder propagated among cannibals in Papua New Guinea through the practice of eating the brain tissue of deceased family members5. Kuru is responsible for first bringing media attention to prion infections in the 1950s5.

Modes of Infection

It is thought that there are at least two ways that human prion diseases are transmitted – through infectious agents from animals (such as ingested infected tissue) or through genetic heritage5. However, there have been recent fears of a third route: through infected tissue transplants (e.g. corneal transplants) or surgical instruments5. In fact, prions cannot be destroyed by boiling, alcohol, acid, standard sterilization methods, or radiation29. Prion-infected brains that have been sitting in formaldehyde for decades can still transmit spongiform disease!29 Standard surgical procedures may have to be modified in the future to accommodate protection from prions.
Koch’s Postulates Revisited
A main reason dissenters remain in the prion debate is that Koch’s postulates – the principles that govern whether an agent is infectious – have not been fulfilled for prion diseases. The main difficulty is that prions cannot be grown in pure culture like bacteria or viruses31. Thus, scientists cannot infect laboratory animals with pure cultures of prions and demonstrate infectiousness in the manner demanded by Koch’s postulates31.
The Final Word?
“Protein, so far as we know, does not replicate itself all by itself, not on this
planet anyway. Looked at this way, the [prion] seems the strangest thing in all
biology and, until someone in some laboratory figures out what it is, a candidate
for Modern Wonder”32.
-Lewis Thomas