
Microbial regulation of intestinal epithelial gene transcription
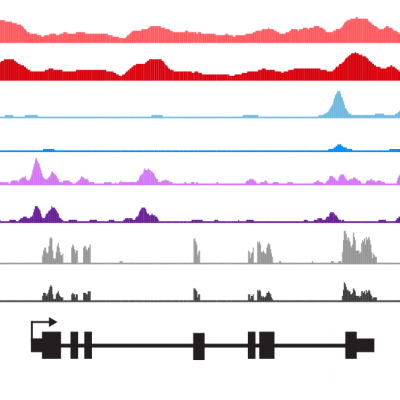 The microbiome has been implicated in an increasing number of disease states in humans and other animals. This has generated significant interest in defining the mechanisms by which host cells perceive and translate signals from the microbiome into specific physiologic and pathophysiologic responses. The intestinal epithelium is the primary interface for host-microbiome interactions, and also serves critical roles in barrier function and nutrient absorption. The mechanisms by which intestinal epithelial cells (IECs) modify their physiology in response to the microbiome remains unclear. Physiologic responses in eukaryotic cells are often mediated by changes in gene transcription that are specific to the cell type and the stimulus. The specificity of the transcriptional response is determined by targeted alterations in chromatin accessibility and post-translational modifications of histone proteins, and consequent interactions between specific transcription factors (TFs) and open chromatin at DNA cis-regulatory regions (CRRs). Previous studies had identified TFs involved in IEC development and inflammatory responses. However, the transcriptional regulatory networks mediating broader IEC responses to the microbiome in the context of health and disease remained almost completely unknown.
The microbiome has been implicated in an increasing number of disease states in humans and other animals. This has generated significant interest in defining the mechanisms by which host cells perceive and translate signals from the microbiome into specific physiologic and pathophysiologic responses. The intestinal epithelium is the primary interface for host-microbiome interactions, and also serves critical roles in barrier function and nutrient absorption. The mechanisms by which intestinal epithelial cells (IECs) modify their physiology in response to the microbiome remains unclear. Physiologic responses in eukaryotic cells are often mediated by changes in gene transcription that are specific to the cell type and the stimulus. The specificity of the transcriptional response is determined by targeted alterations in chromatin accessibility and post-translational modifications of histone proteins, and consequent interactions between specific transcription factors (TFs) and open chromatin at DNA cis-regulatory regions (CRRs). Previous studies had identified TFs involved in IEC development and inflammatory responses. However, the transcriptional regulatory networks mediating broader IEC responses to the microbiome in the context of health and disease remained almost completely unknown.
We are committed to understanding the transcriptional regulatory networks underlying intestinal development and physiology, and how commensal and pathogenic microbes modify these networks in the context of health and disease. Our work was the first to identify host transcriptional responses to microbiome that are conserved between fish and mammals, and to identify initial upstream bacterial signals. Leveraging the strengths of the zebrafish for in vivo analysis of CRR function, we discovered that Hepatocyte nuclear factor 4 (HNF4), an ancient TF family implicated in intestinal physiology and IBD, is a conserved target of microbial regulation. Using our expertise in epigenetics and chromatin biology, we discovered that IEC transcriptional responses to microbiome colonization i n mice are not achieved through changes in chromatin accessibility, but instead through post-translational modifications to histone proteins at microbiome-responsive enhancers. We also recently used a comparative functional genomic approach to uncover an IEC transcriptional regulatory network conserved between fish, mice, and humans and relevant to modern human disorders such as IBD and obesity. These findings empower our current comparative approach to identify specific candidate regulatory mechanisms that govern the developmental identity and microbial sensitivity of IECs in states of health and disease.
n mice are not achieved through changes in chromatin accessibility, but instead through post-translational modifications to histone proteins at microbiome-responsive enhancers. We also recently used a comparative functional genomic approach to uncover an IEC transcriptional regulatory network conserved between fish, mice, and humans and relevant to modern human disorders such as IBD and obesity. These findings empower our current comparative approach to identify specific candidate regulatory mechanisms that govern the developmental identity and microbial sensitivity of IECs in states of health and disease.
Relevant publications:
Rawls, J.F., Samuel, B.S., and Gordon, J.I. (2004) Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proc. Natl. Acad. Sci. U.S.A. 101 (13): 4596-4601. [PMCID: PMC384792]
Rawls, J.F., Mahowald, M.A., Ley, R.E., and Gordon, J.I. (2006) Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell 127(2): 423-433. [PMCID: PMC4839475]
Kanther, M., Sun. S., Mühlbauer, M., Mackey, L.C., Flynn, E.J., Bagnat, M., Jobin, C., and Rawls, J.F. (2011) Microbial colonization induces dynamic temporal and spatial NF-κB responses in the zebrafish digestive tract. Gastroenterology 141(1): 197-207. [PMCID: PMC3164861]
Camp, J.G., Jazwa, A.L., Trent, C.M., and Rawls, J.F. (2012) Intronic cis-regulatory modules mediate tissue-specific and microbial control of angptl4/fiaf transcription. PLoS Genetics 8(3): e1002585. [PMCID: PMC3315460]
Camp, J.G., Frank, C.L., Lickwar, C.R., Guturu, H., Rube, T., Bejerano, G., Crawford, G.E., and Rawls, J.F. (2014) Microbiota modulate transcription in the intestinal epithelium without remodeling the open chromatin landscape. Genome Research 24(9): 1504-16. [PMCID: PMC4158762]
Marjoram. L., Alvers, A., Deerhake, M.E., Bagwell, J., Mankiewicz, J., Cocchiaro, J.L., Beerman, R.W., Willer, J., Katsanis, N., Tobin, D.M., Rawls, J.F., Goll, M., and Bagnat, M. (2015). Epigenetic control of intestinal barrier function and inflammation in zebrafish. Proc Natl Acad Sci USA 112(9): 2770-5. [PMCID: PMC4352795]
Davison, J.M., Lickwar, C.R., Song, L., Breton, G., Crawford, G.E., and Rawls, J.F. (2017) Microbiota regulate intestinal epithelial gene expression by suppressing the transcription factor Hepatocyte nuclear factor 4 alpha. Genome Research 27(7): 1195-1206. [PMCID: PMC5495071]
Lickwar, C.R., Camp, J.G., Weiser, M., Kingsley, D.M., Furey, T.S., Sheikh, S.Z., and Rawls, J.F. (2017) Genomic dissection of conserved transcriptional regulation in intestinal epithelial cells. PLoS Biology 15(8):e2002054. [PMCID: PMC5574553]